
Fused Graphical Lasso Recovers Flowering Time Mutation Genes in Arabidopsis thaliana


Abstract
:1. Introduction
Flowering in Plants
2. Materials and Methods
2.1. Dataset
2.2. Sample Selection Based on Phenotypic Observations
2.3. Data Processing and Filtering
2.4. Gene Selection for Fused Graphical Lasso
2.5. Fused Graphical Lasso Gene Network Inference
2.6. Network Inference Using MARINa Algorithm
3. Results
3.1. Wild Type and Flowering Mutant Subnetworks
3.2. Predicting Mutation Causing Genes
3.3. Predicted Interactions of LNK1/2 Genes
3.4. Comparison with Existing Methods
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abberton, M.; Batley, J.; Bentley, A.; Bryant, J.; Cai, H.; Cockram, J.; Costa de Oliveira, A.; Cseke, L.J.; Dempewolf, H.; De Pace, C.; et al. Global agricultural intensification during climate change: A role for genomics. Plant Biotechnol. J. 2016, 14, 1095–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheben, A.; Yuan, Y.; Edwards, D. Advances in genomics for adapting crops to climate change. Curr. Plant Biol. 2016, 6, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, C.; Iglesius, A.; Yang, X.B.; Epstein, P.R.; Chivian, E. Climate change and extreme weather events-Implications for food production, plant diseases, and pests. Glob. Chang. Hum. Health 2001, 2, 90–104. [Google Scholar]
- Batley, J.; Edwards, D. The application of genomics and bioinformatics to accelerate crop improvement in a changing climate. Curr. Opin. Plant Biol. 2016, 30, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattenberg, P. Agricultural Losses from Winter Storm Exceed $600 Million. Available online: https://today.tamu.edu/2021/03/02/agricultural-losses-from-winter-storm-exceed-600-million/ (accessed on 25 March 2021).
- Nielsen-Gammon, J.W.; Banner, J.L.; Cook, B.I.; Tremaine, D.M.; Wong, C.I.; Mace, R.E.; Gao, H.; Yang, Z.L.; Gonzalez, M.F.; Hoffpauir, R.; et al. Unprecedented drought challenges for Texas water resources in a changing climate: What do researchers and stakeholders need to know? Earths Future 2020, 8, e2020EF001552. [Google Scholar] [CrossRef]
- Pourkheirandish, M.; Golicz, A.A.; Bhalla, P.L.; Singh, M.B. Global role of crop genomics in the face of climate change. Front. Plant Sci. 2020, 11, 922. [Google Scholar] [CrossRef]
- Kole, C.; Muthamilarasan, M.; Henry, R.; Edwards, D.; Sharma, R.; Abberton, M.; Batley, J.; Bentley, A.; Blakeney, M.; Bryant, J.; et al. Application of genomics-assisted breeding for generation of climate resilient crops: Progress and prospects. Front. Plant Sci. 2015, 6, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.K. A cost of disease resistance: Paradigm or peculiarity? Trends Genet. 2003, 19, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Singh, V.K.; Kumar, A.; Powell, W.; Sorrells, M.E. Can genomics deliver climate-change ready crops? Curr. Opin. Plant Biol. 2018, 45, 205–211. [Google Scholar] [CrossRef]
- Korres, N.E.; Norsworthy, J.K.; Tehranchian, P.; Gitsopoulos, T.K.; Loka, D.A.; Oosterhuis, D.M.; Gealy, D.R.; Moss, S.R.; Burgos, N.R.; Miller, M.R.; et al. Cultivars to face climate change effects on crops and weeds: A review. Agron. Sustain. Dev. 2016, 36, 12. [Google Scholar] [CrossRef] [Green Version]
- Craufurd, P.Q.; Wheeler, T.R. Climate change and the flowering time of annual crops. J. Exp. Bot. 2009, 60, 2529–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez, M.A.; Ahn, J.H.; Weigel, D. A thermosensory pathway controlling flowering time in Arabidopsis thaliana. Nat. Genet. 2003, 33, 168–171. [Google Scholar] [CrossRef]
- Jagadish, S.; Bahuguna, R.N.; Djanaguiraman, M.; Gamuyao, R.; Prasad, P.; Craufurd, P.Q. Implications of high temperature and elevated CO2 on flowering time in plants. Front. Plant Sci. 2016, 7, 913. [Google Scholar] [CrossRef] [Green Version]
- Putterill, J.; Laurie, R.; Macknight, R. It’s time to flower: The genetic control of flowering time. Bioessays 2004, 26, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Pineiro, M.; Coupland, G. The control of flowering time and floral identity in Arabidopsis. Plant Physiol. 1998, 117, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, R.; Zeevaart, J.A. Floral stimulus movement in Perilla and flower inhibition caused by noninduced leaves. Plant Physiol. 1973, 51, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Lang, A.; Chailakhyan, M.K.; Frolova, I. Promotion and inhibition of flower formation in a dayneutral plant in grafts with a short-day plant and a long-day plant. Proc. Natl. Acad. Sci. USA 1977, 74, 2412–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irish, E.; Jegla, D. Regulation of extent of vegetative development of the maize shoot meristem. Plant J. 1997, 11, 63–71. [Google Scholar] [CrossRef]
- Pouteau, S.; Nicholls, D.; Tooke, F.; Coen, E.; Battey, N. The induction and maintenance of flowering in Impatiens. Development 1997, 124, 3343–3351. [Google Scholar] [CrossRef]
- Mouradov, A.; Cremer, F.; Coupland, G. Control of flowering time: Interacting pathways as a basis for diversity. Plant Cell 2002, 14, S111–S130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugnone, M.L.; Soverna, A.F.; Sanchez, S.E.; Schlaen, R.G.; Hernando, C.E.; Seymour, D.K.; Mancini, E.; Chernomoretz, A.; Weigel, D.; Más, P.; et al. LNK genes integrate light and clock signaling networks at the core of the Arabidopsis oscillator. Proc. Natl. Acad. Sci. USA 2013, 110, 12120–12125. [Google Scholar] [CrossRef] [Green Version]
- Harmer, S.L.; Hogenesch, J.B.; Straume, M.; Chang, H.S.; Han, B.; Zhu, T.; Wang, X.; Kreps, J.A.; Kay, S.A. Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 2000, 290, 2110–2113. [Google Scholar] [CrossRef] [PubMed]
- Casal, J.J.; Yanovsky, M.J. Regulation of gene expression by light. Int. J. Dev. Biol. 2004, 49, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malapeira, J.; Khaitova, L.C.; Mas, P. Ordered changes in histone modifications at the core of the Arabidopsis circadian clock. Proc. Natl. Acad. Sci. USA 2012, 109, 21540–21545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, T.P.; Mockler, T.C.; Breton, G.; McEntee, C.; Byer, A.; Trout, J.D.; Hazen, S.P.; Shen, R.; Priest, H.D.; Sullivan, C.M.; et al. Network discovery pipeline elucidates conserved time-of-day–specific cis-regulatory modules. PLoS Genet 2008, 4, e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.Y.; Wei, K.C.; Huang, A.C.Y.; Wang, K.; Huang, C.Y.; Yi, D.; Tang, C.Y.; Galas, D.J.; Hood, L.E. RNASEQR—a streamlined and accurate RNA-seq sequence analysis program. Nucleic Acids Res. 2012, 40, e42. [Google Scholar] [CrossRef] [PubMed]
- Lewy, A.; Emens, J.; Songer, J.; Rough, J. The neurohormone melatonin as a marker, medicament, and mediator. In Hormones, Brain and Behavior Online; Elsevier Inc.: Amsterdam, The Netherlands, 2010; pp. 2505–2528. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Huala, E.; Dickerman, A.W.; Garcia-Hernandez, M.; Weems, D.; Reiser, L.; LaFond, F.; Hanley, D.; Kiphart, D.; Zhuang, M.; Huang, W.; et al. The Arabidopsis Information Resource (TAIR): A comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 2001, 29, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Bolser, D.; Staines, D.M.; Pritchard, E.; Kersey, P. Ensembl plants: Integrating tools for visualizing, mining, and analyzing plant genomics data. In Plant Bioinformatics; Springer: Berlin, Germany, 2016; pp. 115–140. [Google Scholar]
- Lahiri, A.; Zhou, L.; He, P.; Datta, A. Detecting Drought Regulators using Stochastic Inference in Bayesian Networks. 2020. [Google Scholar]
- Lahiri, A.; Venkatasubramani, P.S.; Datta, A. Bayesian modeling of plant drought resistance pathway. BMC Plant Biol. 2019, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Arshad, O.A.; Datta, A. Towards targeted combinatorial therapy design for the treatment of castration-resistant prostate cancer. BMC Bioinform. 2017, 18, 5–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, R.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M.L. A Gaussian Mixture-Model Exploiting Pathway Knowledge for Dissecting Cancer Heterogeneity. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 17, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Saraf, R.S.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M. An in-silico study examining the induction of apoptosis by Cryptotanshinone in metastatic melanoma cell lines. BMC Cancer 2018, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saraf, R.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M.L.; Miller, T.; Wilson-Robles, H.M. In silico modeling of the induction of apoptosis by Cryptotanshinone in osteosarcoma cell lines. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Vundavilli, H.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M. Targeting oncogenic mutations in colorectal cancer using cryptotanshinone. PLoS ONE 2021, 16, e0247190. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, T.; González, B.; Ruz, G.A. Reconstruction of a gene regulatory network of the induced systemic resistance defense response in Arabidopsis using boolean networks. BMC Bioinform. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkat, P.S.; Narayanan, K.R.; Datta, A. A Bayesian network-based approach to selection of intervention points in the mitogen- activated protein kinase plant defense response pathway. J. Comput. Biol. 2017, 24, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Vijesh, N.; Chakrabarti, S.K.; Sreekumar, J. Modeling of gene regulatory networks: A review. J. Biomed. Sci. Eng. 2013, 6, 223. [Google Scholar] [CrossRef] [Green Version]
- Vundavilli, H.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M. Bayesian inference identifies combination therapeutic targets in breast cancer. IEEE Trans. Biomed. Eng. 2019, 66, 2684–2692. [Google Scholar] [CrossRef]
- Vundavilli, H.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M. Using Chou’s 5-steps rule to Model Feedback in Lung Cancer. IEEE J. Biomed. Health Inform. 2019, 24, 2430–2438. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Wang, P.; Witten, D.M. The joint graphical lasso for inverse covariance estimation across multiple classes. J. R. Stat. Society. Ser. B Stat. Methodol. 2014, 76, 373. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.; Parikh, N.; Chu, E. Distributed Optimization and Statistical Learning Via the Alternating Direction Method of Multipliers. Mach. Learn. 2010, 3, 1–122. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rajbhandari, P.; Alvarez, M.J.; Bandaru, P.; Lim, W.K.; Sato, M.; Wang, K.; Sumazin, P.; Kustagi, M.; Bisikirska, B.C.; et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 2010, 6, 377. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, A.; Giorgi, F.M.; Lopez, G.; Califano, A. ARACNe-AP: Gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 2016, 32, 2233–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, S.; Oda, A.; Yoshida, R.; Niinuma, K.; Miyata, K.; Tomozoe, Y.; Tajima, T.; Nakagawa, M.; Hayashi, K.; Coupland, G.; et al. Circadian clock proteins LHY and CCA1 regulate SVP protein accumulation to control flowering in Arabidopsis. Plant Cell 2008, 20, 2960–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.C.; Lasswell, J.; Rogg, L.E.; Cohen, M.A.; Bartel, B. FKF1, a clock-controlled gene that regulates the transition to flowering in Arabidopsis. Cell 2000, 101, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Sawa, M.; Nusinow, D.A.; Kay, S.A.; Imaizumi, T. FKF1 and GIGANTEA complex formation is required for day-length measurement in Arabidopsis. Science 2007, 318, 261–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ma, D.; Lu, S.X.; Hu, X.; Huang, R.; Liang, T.; Xu, T.; Tobin, E.M.; Liu, H. Blue light-and low temperature-regulated COR27 and COR28 play roles in the Arabidopsis circadian clock. Plant Cell 2016, 28, 2755–2769. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Li, C.; Xu, S.; Xing, L.; Xu, Y.; Chong, K. JACALIN-LECTIN LIKE1 regulates the nuclear accumulation of GLYCINE-RICH RNA-BINDING PROTEIN7, influencing the RNA processing of FLOWERING LOCUS C antisense transcripts and flowering time in Arabidopsis. Plant Physiol. 2015, 169, 2102–2117. [Google Scholar] [PubMed] [Green Version]
- Streitner, C.; Danisman, S.; Wehrle, F.; Schöning, J.C.; Alfano, J.R.; Staiger, D. The small glycine-rich RNA binding protein AtGRP7 promotes floral transition in Arabidopsis thaliana. Plant J. 2008, 56, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Lee, S.Y.; Seok, H.Y.; Kim, S.H.; Sung, Z.R.; Moon, Y.H. EMF1 interacts with EIP1, EIP6 or EIP9 involved in the regulation of flowering time in Arabidopsis. Plant Cell Physiol. 2011, 52, 1376–1388. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Jiang, H.W.; Hsieh, E.J.; Chen, H.Y.; Chien, C.T.; Hsieh, H.L.; Lin, T.P. Drought and salt stress tolerance of an Arabidopsis glutathione S-transferase U17 knockout mutant are attributed to the combined effect of glutathione and abscisic acid. Plant Physiol. 2012, 158, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleet, C.; Yen, J.; Hill, E.; Gillaspy, G. Co-suppression of AtMIPS demonstrates cooperation of MIPS1, MIPS2 and MIPS3 in maintaining myo-inositol synthesis. Plant Mol. Biol. 2018, 97, 253–263. [Google Scholar] [CrossRef]
- Krahmer, J.; Goralogia, G.S.; Kubota, A.; Zardilis, A.; Johnson, R.S.; Song, Y.H.; MacCoss, M.J.; Le Bihan, T.; Halliday, K.J.; Imaizumi, T.; et al. Time-resolved interaction proteomics of the GIGANTEA protein under diurnal cycles in Arabidopsis. FEBS Lett. 2019, 593, 319–338. [Google Scholar] [CrossRef] [Green Version]
- Kou, X.; Watkins, C.B.; Gan, S.S. Arabidopsis AtNAP regulates fruit senescence. J. Exp. Bot. 2012, 63, 6139–6147. [Google Scholar] [CrossRef] [Green Version]
- Son, O.; Hur, Y.S.; Kim, Y.K.; Lee, H.J.; Kim, S.; Kim, M.R.; Nam, K.H.; Lee, M.S.; Kim, B.Y.; Park, J.; et al. ATHB12, an ABA- inducible homeodomain-leucine zipper (HD-Zip) protein of Arabidopsis, negatively regulates the growth of the inflorescence stem by decreasing the expression of a gibberellin 20-oxidase gene. Plant Cell Physiol. 2010, 51, 1537–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.F.; Wang, Z.Y. Overexpression of COL9, a CONSTANS-LIKE gene, delays flowering by reducing expression of CO and FT in Arabidopsis thaliana. Plant J. 2005, 43, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Carvallo, M.; Hamilton, E.E.; Preuss, S.; Kay, S.A. Arabidopsis B-BOX32 interacts with CONSTANS-LIKE3 to regulate flowering. Proc. Natl. Acad. Sci. USA 2017, 114, 172–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Hu, H.; Ren, H.; Kong, Y.; Lin, H.; Guo, J.; Wang, L.; He, Y.; Ding, X.; Grabsztunowicz, M.; et al. LIGHT-INDUCED RICE1 regulates light-dependent attachment of LEAF-TYPE FERREDOXIN-NADP+ OXIDOREDUCTASE to the thylakoid membrane in rice and Arabidopsis. Plant Cell 2016, 28, 712–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciannamea, S.; Jensen, C.S.; Agerskov, H.; Petersen, K.; Lenk, I.; Didion, T.; Immink, R.G.; Angenent, G.C.; Nielsen, K.K. A new member of the LIR gene family from perennial ryegrass is cold-responsive, and promotes vegetative growth in Arabidopsis. Plant Sci. 2007, 172, 221–227. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamichi, N. The Transcriptional Network in the Arabidopsis Circadian Clock System. Genes 2020, 11, 1284. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wang, P.; Liu, X.; Yuan, L.; Wang, L.; Zhang, C.; Li, Y.; Xing, H.; Zhi, L.; Yue, Z.; et al. LNK1 and LNK2 are transcriptional coactivators in the Arabidopsis circadian oscillator. Plant Cell 2014, 26, 2843–2857. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.S.; Imaizumi, T. Circadian clock and photoperiodic response in Arabidopsis: From seasonal flowering to redox homeostasis. Biochemistry 2015, 54, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.Y.; Park, S.; Lee, S.; Lee, S.S.; Imaizumi, T.; Song, Y.H. GIGANTEA regulates the timing stabilization of CONSTANS by altering the interaction between FKF1 and ZEITLUPE. Mol. Cells 2019, 42, 693. [Google Scholar]
- Song, Y.H.; Estrada, D.A.; Johnson, R.S.; Kim, S.K.; Lee, S.Y.; MacCoss, M.J.; Imaizumi, T. Distinct roles of FKF1, GIGANTEA, and ZEITLUPE proteins in the regulation of CONSTANS stability in Arabidopsis photoperiodic flowering. Proc. Natl. Acad. Sci. USA 2014, 111, 17672–17677. [Google Scholar] [CrossRef] [Green Version]


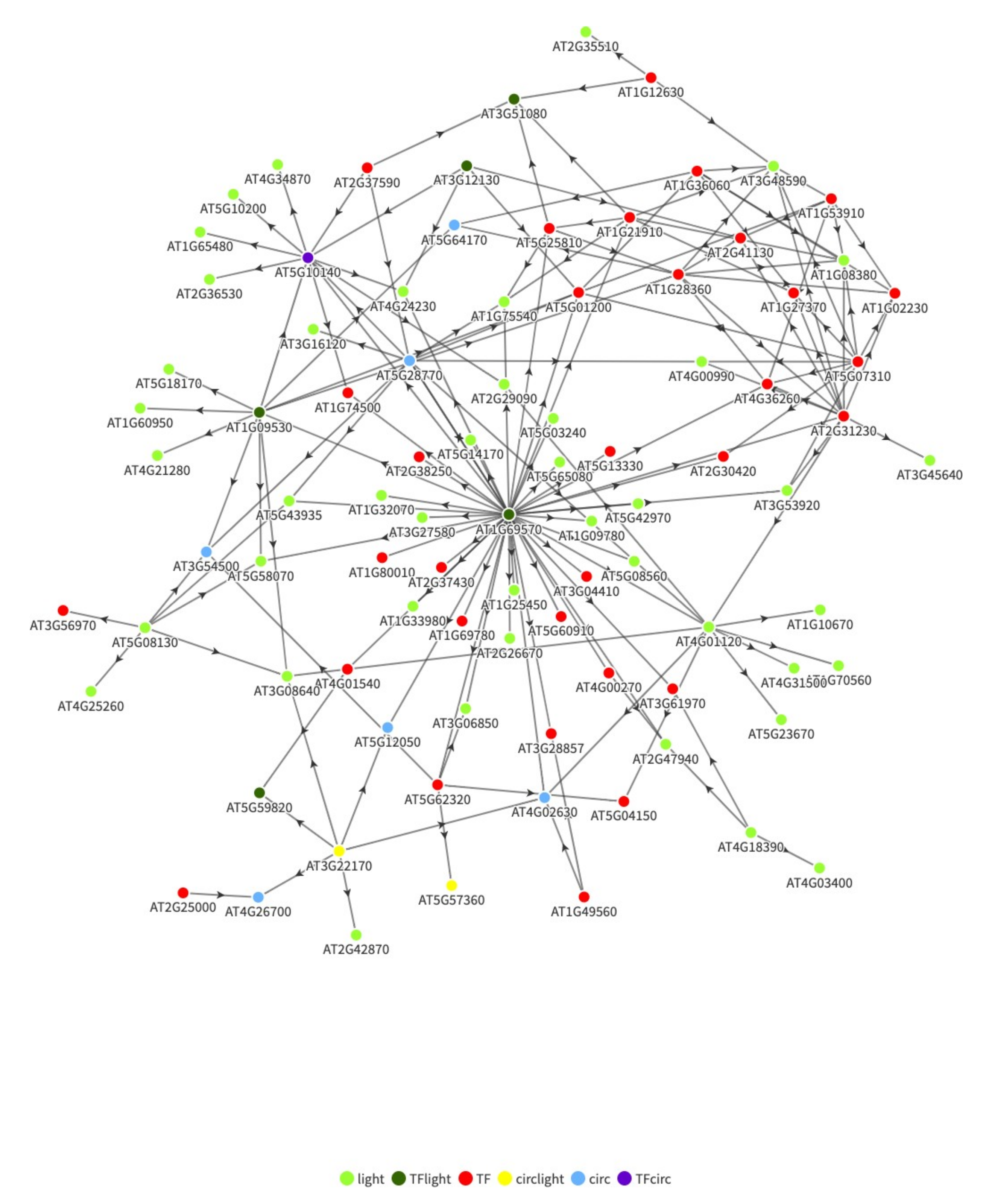{kind=link}
| Topological Properties | WT Subnetwork | Mutant Subnetwork |
|---|---|---|
| Number of subnetworks in each class | 1 | 1 |
| Number of edges in each class | 728 | 92 |
| Degree LNK1 gene (AT5G64170) | 5 | 0 |
| Degree LNK2 gene (AT3G54500) | 18 | 0 |
| Topological Properties | WT Hub | Gene Name | #edges | Mutant Hub | Gene Name | #edges |
|---|---|---|---|---|---|---|
| 1 | AT2G46830 | CCA1 | 155 | AT1G01060 | LHY | 70 |
| 2 | AT1G01060 | LHY | 153 | AT2G46830 | CCA1 | 12 |
| 3 | AT1G68050 | FKF1 | 43 | AT3G26740 | CCL | 12 |
| 4 | AT5G42900 | COR27 | 43 | AT1G01520 | RVE3 | 4 |
| 5 | AT1G73870 | COL7 | 40 | AT1G69490 | NAP | 4 |
| 6 | AT2G21660 | GRP7 | 39 | AT1G10370 | ATGSTU17 | 3 |
| 7 | AT3G12320 | LNK3 | 37 | AT3G21150 | EIP6,BBOX32 | 3 |
| 8 | AT3G26740 | CCL | 32 | AT3G61890 | ATHB12 | 3 |
| 9 | AT1G07180 | NDA1 | 29 | AT1G26790 | CDF6 | 2 |
| 10 | AT5G60100 | PRR3 | 27 | AT2G22240 | MIPS2 | 2 |
| INTERPRO ID | Description | #Genes | Strength | FDR |
|---|---|---|---|---|
| IPRO39928 | LNK family | 4 of 4 | 2.34 | |
| IPRO24708 | Catalase active site | 2 of 2 | 2.34 | 0.0015 |
| IPRO24711 | Catalase, mono-functional, haem-containing clades 1 and 3 | 2 of 3 | 2.16 | 0.0023 |
| IPRO20835 | Catalase superfamily | 2 of 3 | 2.16 | 0.0023 |
| IPRO18028 | Catalase, mono-functional, haem-containing | 2 of 3 | 2.16 | 0.0023 |
| IPRO11614 | Catalase core domain | 2 of 3 | 2.16 | 0.0023 |
| IPRO10582 | Catalase immune-responsive domain | 2 of 3 | 2.16 | 0.0023 |
| IPRO002226 | Catalase haem-binding site | 2 of 3 | 2.16 | 0.0023 |
| IPRO039615 | Protein PHYTOCHROME KINASE SUBSTRATE | 2 of 4 | 2.04 | 0.0026 |
| IPRO000315 | B-boc type zinc finger | 14 of 34 | 1.95 |
| Gene ID | Name | Strength |
|---|---|---|
| AT1G07180 | NDA1 | −0.05559 |
| AT1G10370 | ATGSTU17 | −0.03301 |
| AT1G68050 | FKF1 | 0.06481 |
| AT1G73870 | COL7 | −0.07189 |
| AT2G21130 | CYP19-2 | 0.00361 |
| AT2G21660 | GRP7 | 0.04804 |
| AT2G40080 | ELF4 | 0.05177 |
| AT2G46830 | CCA1 | −0.07997 |
| AT3G02380 | COL2 | −0.03379 |
| AT3G07650 | COL9 | 0.05028 |
| AT3G12320 | LNK3 | −0.06966 |
| AT3G20810 | JMJD5 | 0.04070 |
| AT4G33980 | COR28 | 0.04301 |
| AT5G06980 | LNK4 | −0.04090 |
| AT5G24120 | SIGE | −0.02336 |
| AT5G42900 | COR27 | 0.00917 |
| AT5G48250 | BBX8 | 0.00460 |
| AT5G60100 | PRR3 | 0.04671 |
| Gene ID | Name | Strength |
|---|---|---|
| AT1G68050 | FKF1 | 0.05274 |
| AT1G73870 | COL7 | −0.04299 |
| AT2G21660 | GRP7 | 0.00164 |
| AT2G46830 | CCA1 | −0.29688 |
| AT3G12320 | LNK3 | −0.03224 |
| Rank | Master Regulator | Size | NES | p-Value | FDR |
|---|---|---|---|---|---|
| 1 | FLC | 48 | 3.51 | 0.000452 | 0.291 |
| 2 | GT-3b | 25 | 3.34 | 0.000829 | 0.291 |
| 3 | ERF12 | 29 | 3.18 | 0.001460 | 0.291 |
| 4 | AT4G39160 | 49 | 3.13 | 0.001740 | 0.291 |
| 5 | PIF3 | 35 | 3.05 | 0.002250 | 0.314 |
| 6 | SRS2 | 92 | 3.00 | 0.002690 | 0.315 |
| 7 | ERF115 | 42 | 2.98 | 0.002880 | 0.315 |
| 8 | NAM | 34 | −3.12 | 0.001830 | 0.291 |
| 9 | BHLH038 | 65 | −3.15 | 0.001620 | 0.291 |
| 10 | NGA2 | 63 | −3.47 | 0.000529 | 0.291 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapoor, R.; Datta, A.; Thomson, M. Fused Graphical Lasso Recovers Flowering Time Mutation Genes in Arabidopsis thaliana. Inventions 2021, 6, 52. https://doi.org/10.3390/inventions6030052
Kapoor R, Datta A, Thomson M. Fused Graphical Lasso Recovers Flowering Time Mutation Genes in Arabidopsis thaliana. Inventions. 2021; 6(3):52. https://doi.org/10.3390/inventions6030052
Chicago/Turabian StyleKapoor, Rajan, Aniruddha Datta, and Michael Thomson. 2021. "Fused Graphical Lasso Recovers Flowering Time Mutation Genes in Arabidopsis thaliana" Inventions 6, no. 3: 52. https://doi.org/10.3390/inventions6030052