1. Introduction
The foundations of molecular electronics began establishment in the 1970s when Aviram and Ratner prepared and characterized a molecular system with a donor–acceptor species by performing electron transfer tests, in order to find devices with rectifying properties when the system was submitted to a potential difference [
1]. In the last decades, the interest in the study of low dimensional molecular systems has increased in search of their possible use and application. In particular, systems such as organic molecules have been analyzed by coupling them to electrodes, obtaining conductive, semiconducting, and/or insulating behaviors, making them worthy of being considered for electronic connectors, rectifiers, amplifiers, and/or storage devices [
2,
3,
4,
5].
Molecular systems have sparked great interest in the field of optoelectronic devices due to their unique properties when interacting with light. These systems, which can consist of individual molecules or molecular assemblies, exhibit exceptional capabilities to absorb, emit, or scatter photons. This light-interacting characteristic of molecular systems has driven their application in the field of renewable energy, particularly in the development of solar cells. Solar cells based on molecular systems offer a sustainable solution to the growing energy demand by harnessing solar light, an unlimited and environmentally friendly energy source. The efficient conversion of sunlight and waste heat into usable electricity presents a fundamental challenge for researchers in this field. The goal is to fully replace the use of fossil fuels, thereby avoiding environmental pollution and promoting a cleaner and more sustainable means of energy generation [
6,
7,
8,
9].
Molecular systems can exhibit nonlinear optical properties, which can be utilized in applications such as optical information processing, integrated optics, and telecommunications. Compounds with large delocalized electron systems show exceptionally large nonlinear responses and higher laser damage thresholds compared to inorganic materials. Furthermore, these properties can be modified to optimize additional properties, such as mechanical and thermal stability. An example of such molecular compounds with a significantly large first-order nonlinear optical response is diphenyl-ether [
10].
According to the above, in the literature, we find very interesting analyses of thermoelectric and magnetic properties in molecular systems, such as DNA chains, benzene molecules, biphenyl molecules [
11,
12,
13,
14], and especially theoretical-experimental studies of these properties through the molecular system diphenyl-ether. Motivated by the particular characteristics of this last diphenyl-ether molecular system, especially those reported by Dadosh et al. (2005), where they characterized it as a conducting molecular device, we analyze its thermoelectric properties, taking into account different structural configurations.
To complement the above, it is important to emphasize that Dadosh et al. studied a system of three short organic molecules: 4,4-biphenyldithiol (BPD) conjugated molecule; a bis-(4-mercaptophenyl)-ether (BPE) molecule, in which the conjugate is broken in the center by an oxygen atom; and the 1,4-benzenedimethanethiol (BDMT) molecule, in which the conjugate is broken near the contacts by a methylene group, concluding that the oxygen in BPE and the methylene groups in BDMT suppress electrical conduction relative to BPD [
15]. Now, to make our study of the diphenyl-ether properties even more interesting, we highlight the work by S.K. Maiti, who performed a theoretical study of the electron transport properties through single conjugated molecules (BPD, BPE, and BDMT) sandwiched between two non-superconducting electrodes, using the Green’s function technique, within a tight-binding model, finding that the electron transport properties are significantly influenced by the existence of localized groups in these conjugated molecules and the coupling strength between the molecule and the electrode [
16].
On the other hand, we know that to analyze any system of low dimensionality—leading to a characterization through electrical, thermal, spintronic, structural properties, among others—there are many methods used both theoretically and experimentally. For example, at the experimental level, we find the mechanically controlled breakage joints (MCBJ), which search for the relationship of the electrical conductance as a function of the electrode separation, the scanning tunneling microscope (STM), which seeks to characterize the tunneling current with the application of a voltage [
17,
18,
19,
20,
21,
22,
23,
24,
25], among others. On the side of theoretical methods, we find, among many, two important ways: one (strictly numerical) that includes the application of quantum correlation functional theory through effective equations of a single particle, also known as the density functional theory (DFT), and another (numerical and/or analytical) that includes a process of renormalization of the real space by using Green’s functions, where the degrees of freedom of the system are reduced to obtain a one-dimensional system that contains all the structural information of the quantum system [
26,
27,
28,
29,
30].
It is important to remark that with the two theoretical methods mentioned above, it is possible to determine both the electrical and thermal properties, as well as the spintronic properties of different molecular systems; however, in this work we have used the second one (renormalization process) due to its low computational cost compared to the first one (DFT), facilitating even more numerical or analytical calculations [
31,
32,
33,
34,
35,
36].
Returning to the purpose of this work, it should be noted that we focus on determining the thermoelectric properties of the planar diphenyl-ether molecule, taking into account different structural forms, which can provide interesting behaviors such as obtaining a high figure of merit (
), which depends on the electrical conductance (
), the Seebeck coefficient (
S), and the thermal conductance (
). Now, to obtain a more extensive knowledge of the
behavior, these thermoelectric quantities are explored using the already known and standard method of tight binding, calculating the transmission probability (
T) by Green’s functions through the Fisher–Lee relation [
37,
38,
39], while the thermoelectric quantities are evaluated by Landauer’s theory [
38,
39].
Our paper is organized as follows. In
Section 2, we present the model of the diphenyl-ether molecule based on a TB Hamiltonian. In
Section 3, we describe the method used for the calculations of different thermoelectric quantities. Finally, we report our essential discoveries as conclusions in
Section 5. In addition to this, an
Appendix A is presented as a complement to explain in detail the renormalization method or process of the system under study.
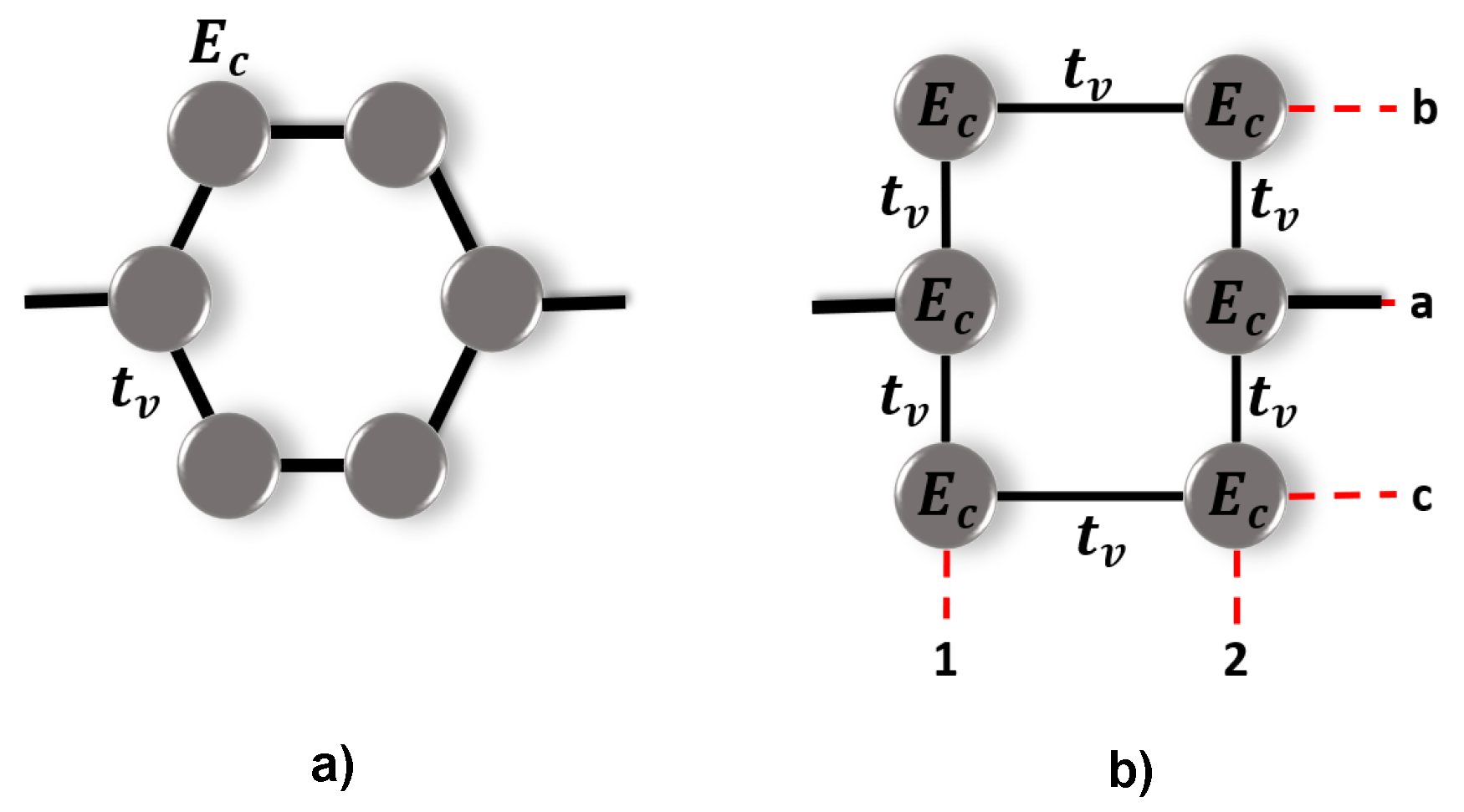
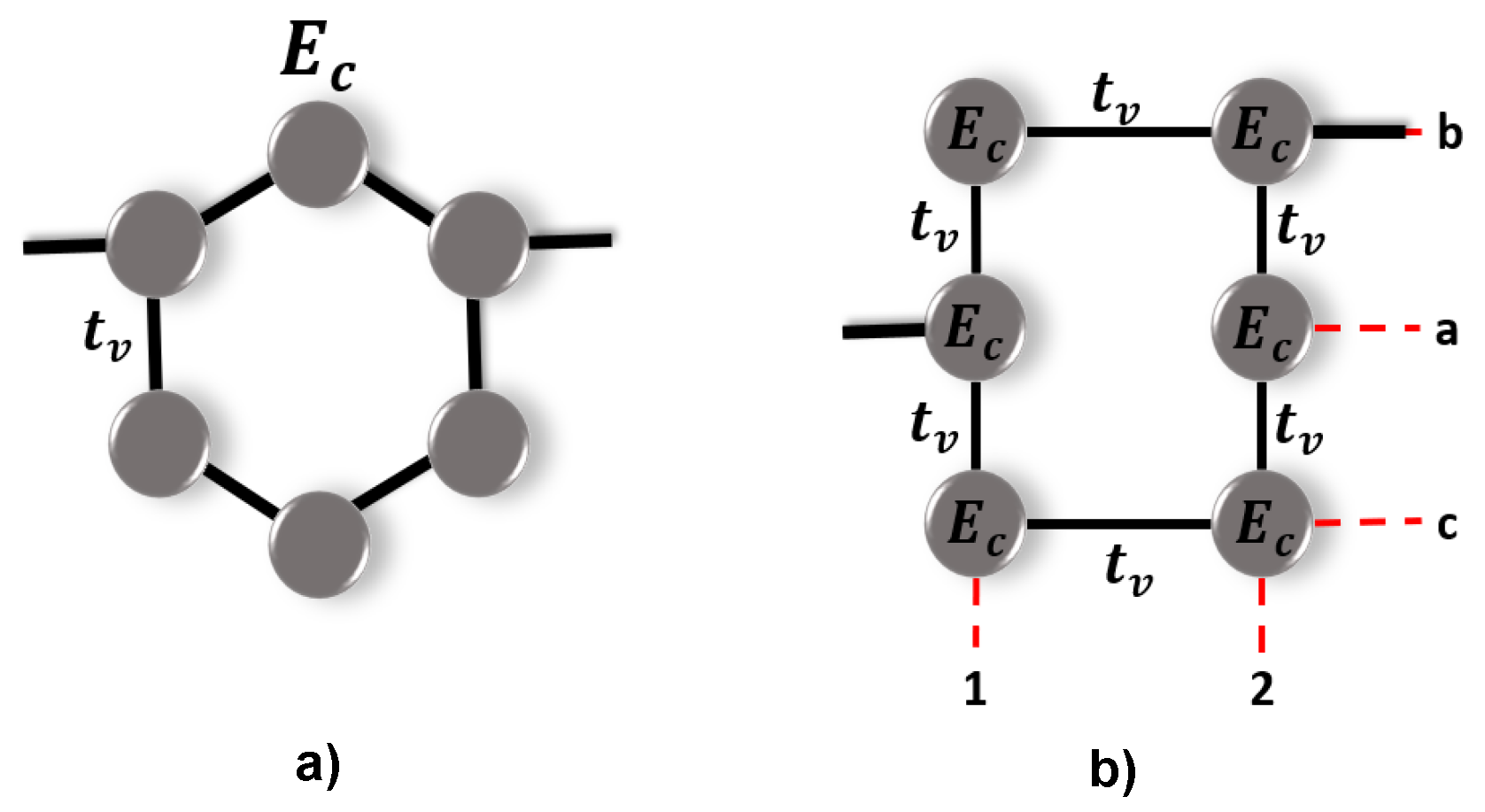
2. Model
To study the transport properties of the diphenyl ether molecule (C
H
O), it is connected between two metal electrodes (Left-
L and Right-
R) through the atomic site
i, resulting in different structural configurations depending on the connections between
, as shown in
Figure 1. In the tight-binding approximation, the total system can be represented by the Hamiltonian given by:
where
corresponds to the Hamiltonian of the molecule, which has the following form:
where
and
are the operators for creating and destroying an electron at site
i.
is the site energy for the carbon (
) or oxygen (
) atoms,
is the hopping between the atoms, which can be
for C−C coupling into the benzene molecule, and
, when the coupling is between the benzene molecules and the oxygen atom. The terms
and
of Equation (
1) represent the Hamiltonian of the leads and the molecule-leads interaction, respectively, and are given by:
here, the operator
is the creation operator of an electron in a state
with energy
;
is the coupling between each lead with the molecule, and
h.c. is the complex conjugate of the Hamiltonian.
3. Method
To determine the thermoelectric properties of the diphenyl-ether molecule, we first focus on calculating the fundamental property that we need in order to determine the others, which is defined as the transmission probability
as a function of the energy with which the electron enters through the molecular system. This property is calculated using Green’s function techniques, through the Fisher–Lee relation [
37,
38,
39] and is given by:
where
represents the advanced (retarded) Green’s functions and
represents the spectral density matrices of the Left (Right) electrodes, which are given by
(were
). Now, to calculate the total Green functions given in the expression (
5) for each model (see
Appendix A for details on the calculation of Green’s functions for each model using the renormalization scheme), the Dyson equation is used, taking into account that
, where
is the Green function of the molecular system without being coupled to the contacts. It is important to note that, as soon as the decimation process is performed for all models, and the systems are reduced to one-dimensional models, the Fisher–Lee relation for our calculations becomes:
where
N is the number of sites in the new linear system.
Then, once the transmission probability is determined by relationship (
6), we can analyze the antiresonances in the
profile for all models, where both the Green’s function and transmission function are equal to zero, indicating that the cofactor is also zero. However, not all zeros of the cofactor result in an antiresonance in the transmission; therefore, we calculate the cofactor where there is no antiresonance and are able to calculate the Green’s function, as a function of the cofactor, using the expression:
where
is the cofactor of the hamiltonian matrix
H and is given by:
where
is a submatrix of
H obtained by removing the
jth row and the
ith column [
40].
In particular, when there is a cyclic molecular system, such as a benzene ring, it is possible to convert the 6-site system to two effective sites, with a
effective Hamiltonian matrix having the following form:
where the diagonal terms (
and
) are the effective energies of sites
a and
b, respectively, which result from renormalizing the
with energy
to an effective site, and off-diagonal terms are the effective couplings between the two effective sites mentioned; with this Hamiltonian, we calculate the Green’s function from site
a to
b with Equation (
7), given the expression:
From Equation (
10), we can observe that when
is zero, the Green’s function is zero, and therefore, the profile in the transmission probability results in an anti-resonance [
41].
Likewise, having the transmission calculated, the current flowing through the molecular system (which is considered as a process of dispersion of an electron between the contacts), is calculated using the Landauer formalism by means of the expression:
where
,
e is the electronic charge,
h represents the Plank’s constant, and
is the Fermi–Dirac distribution function, where
is the Botzmann’s factor, and
is the chemical potential [
41].
Additionally, we use the Landauer integrals
, which have the following expression:
where
describes the equilibrium Fermi energy of the system under the zero bias condition. With this expression (Equation (
7)), we calculate the thermoelectric properties (object of this work) such as the electrical conductance
, the Seebeck coefficient
S, the thermal conductance
, and the figure of merit
defined as:
here,
is the equilibrium temperature [
25].
4. Results and Discussion
In this section, we analyze the quantum transport properties (
,
,
,
S,
, and
) through a single diphenyl-ether molecule, considering four different structural models (see
Figure 1).
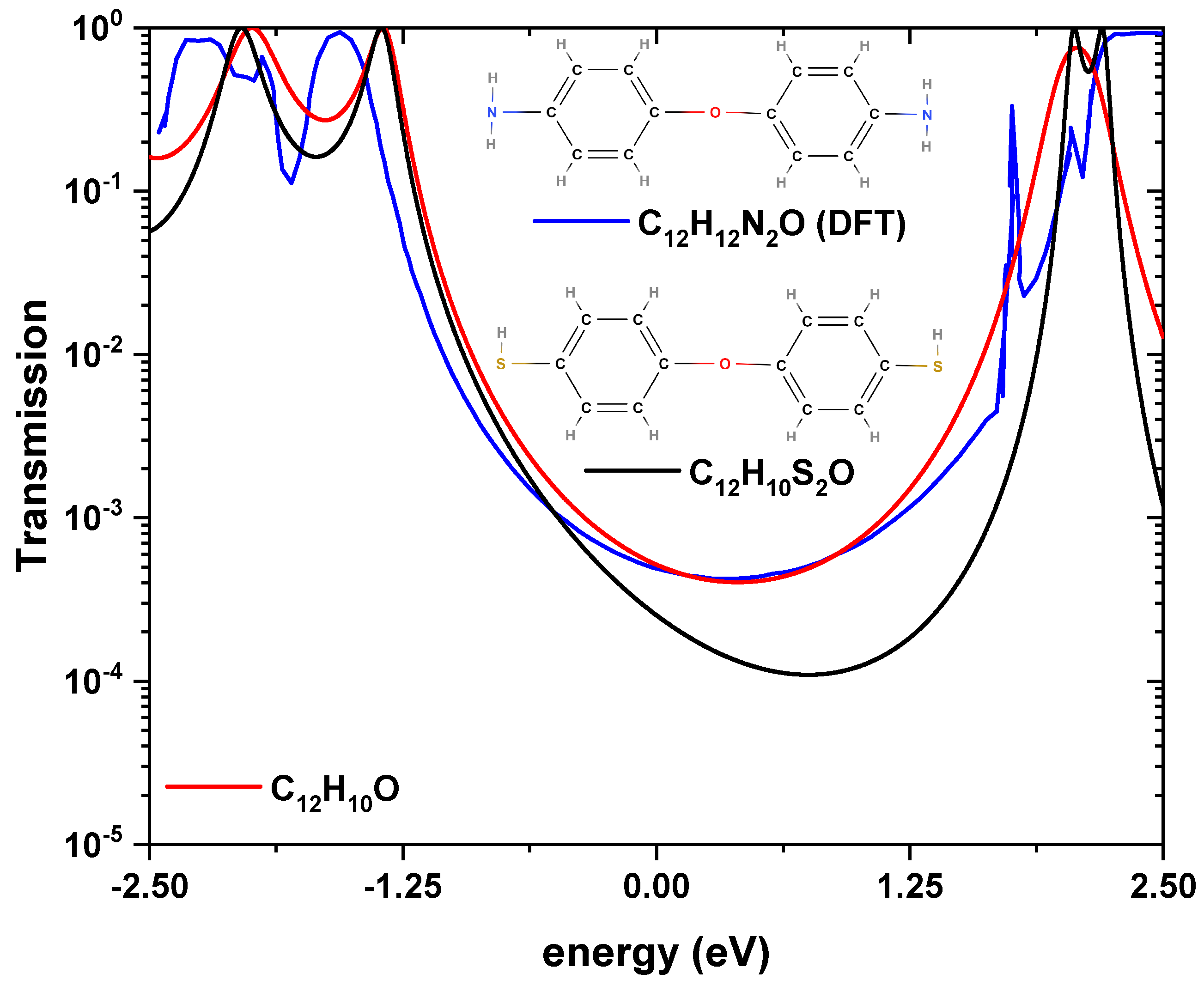
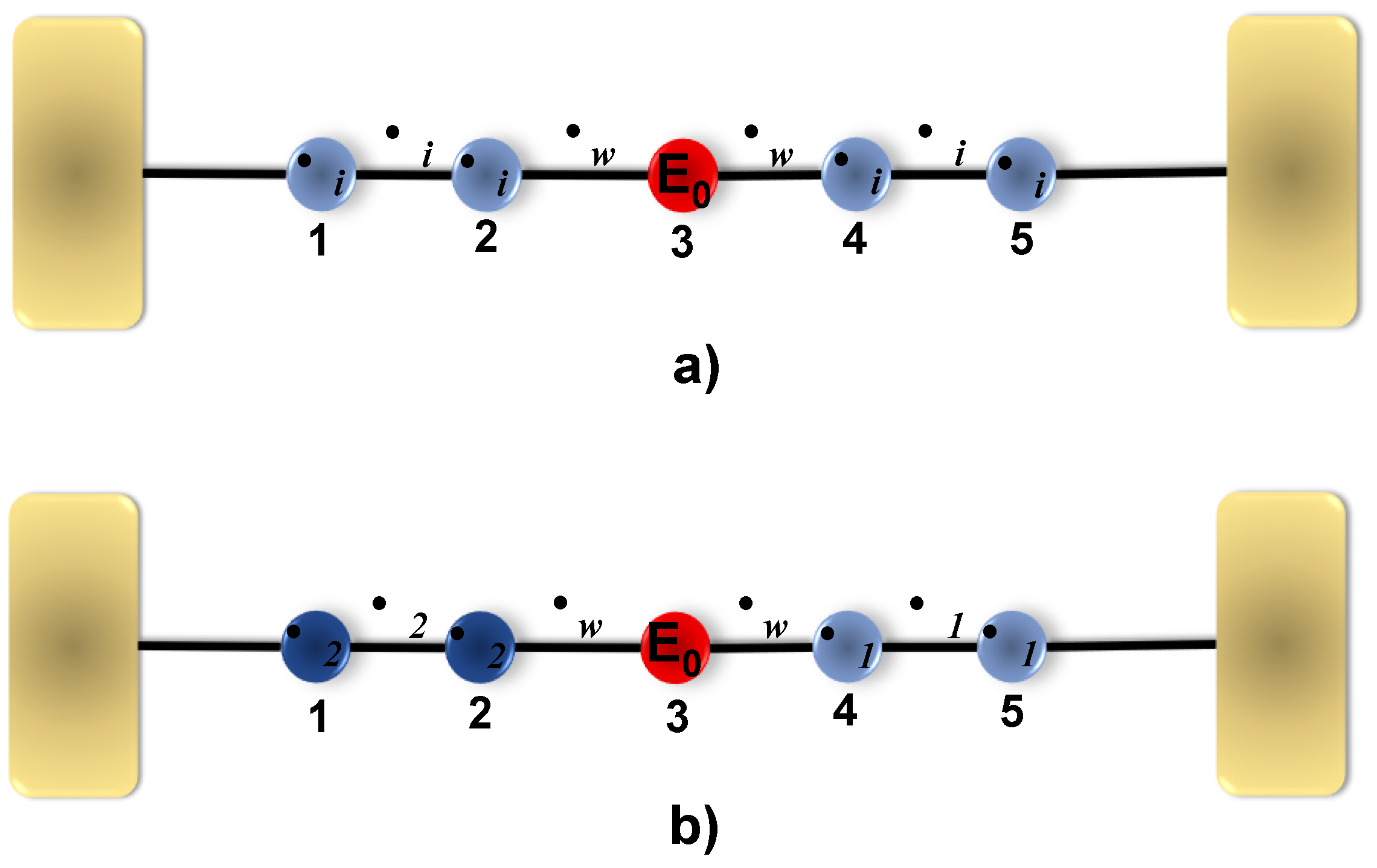
In the first instance, and in order to validate the real space renormalization method used in this work, a comparison of the calculation of the transmission probability determined for two molecular systems similar to that of diphenyl-ether is performed (see
Figure 2). The first system taken into account is a 4,4
-diaminodiethyl diphenyl-ether molecule (C
H
N
O), through which Wang Y.H et al. calculated the transmission using the DFT method. Such a molecule contains two benzene rings linked by an oxygen atom, two NH
groups that couple the molecule to the electrodes, and a second system that is also characterized by two benzene rings with two H atoms, which serve as a bridge to couple the molecule to the electrodes and is called bis-(4-mercaptophenyl)-ether (C
H
S
O). For this last molecular system, the transmission is calculated analytically with the method used in this work, where the site energy used for the Sulfur atom
eV, the site energy of the Carbon atom
eV, the site energy for the Oxygen atom
eV. For the hopping values, we have S–C,
eV; C–C,
eV; and O–C, is
eV.
Figure 2 shows the probability of transmission as a function of the energy of the incident electron, for the 4,4
-diaminodiphenyl-ether systems calculated by DFT (blue curve), and the bis-(4-mercaptophenyl)-ether molecule by the analytical method (black curve), which is compared with the data obtained for the diphenyl-ether model (a) system (red curve). We observed that around the Fermi level (
), the same behavior occurs, which is a destructive quantum interference in the transmission caused by the oxygen site, which breaks the delocalization of the system; this agrees with results from previous works [
15]. On the other hand,
Figure 2 shows that for the three systems, the energy of the HOMO and the LUMO are very similar, which are around −1.25 and 2.00 eV, presenting a gap of approximately the same width. In order to find a good comparison with the system reported by DFT, a
eV was used for the bis-(4-mercaptophenyl)-ether system, and a
eV for the dyphenyl-ether system, which tells us that the additional radicals cause a stronger coupling with the electrodes, compared to the dyphenyl-ether molecule alone. It is important to note that the
-parameter for the three molecules is different,
eV corresponds to a strong coupling to the electrodes where the electrode molecular orbitals hybridize strongly with the molecule orbitals, while
eV corresponds to a weaker hybridization. In the DFT calculation, no report of
was expelled. Furthermore, the magnitude difference around the Fermi level for the three curves was irrelevant since it accounts for very small transmission values. In the models shown in
Figure 1, the site energies
eV and
eV were taken, as the potential between the C–C sites is
eV and C–O is
eV.
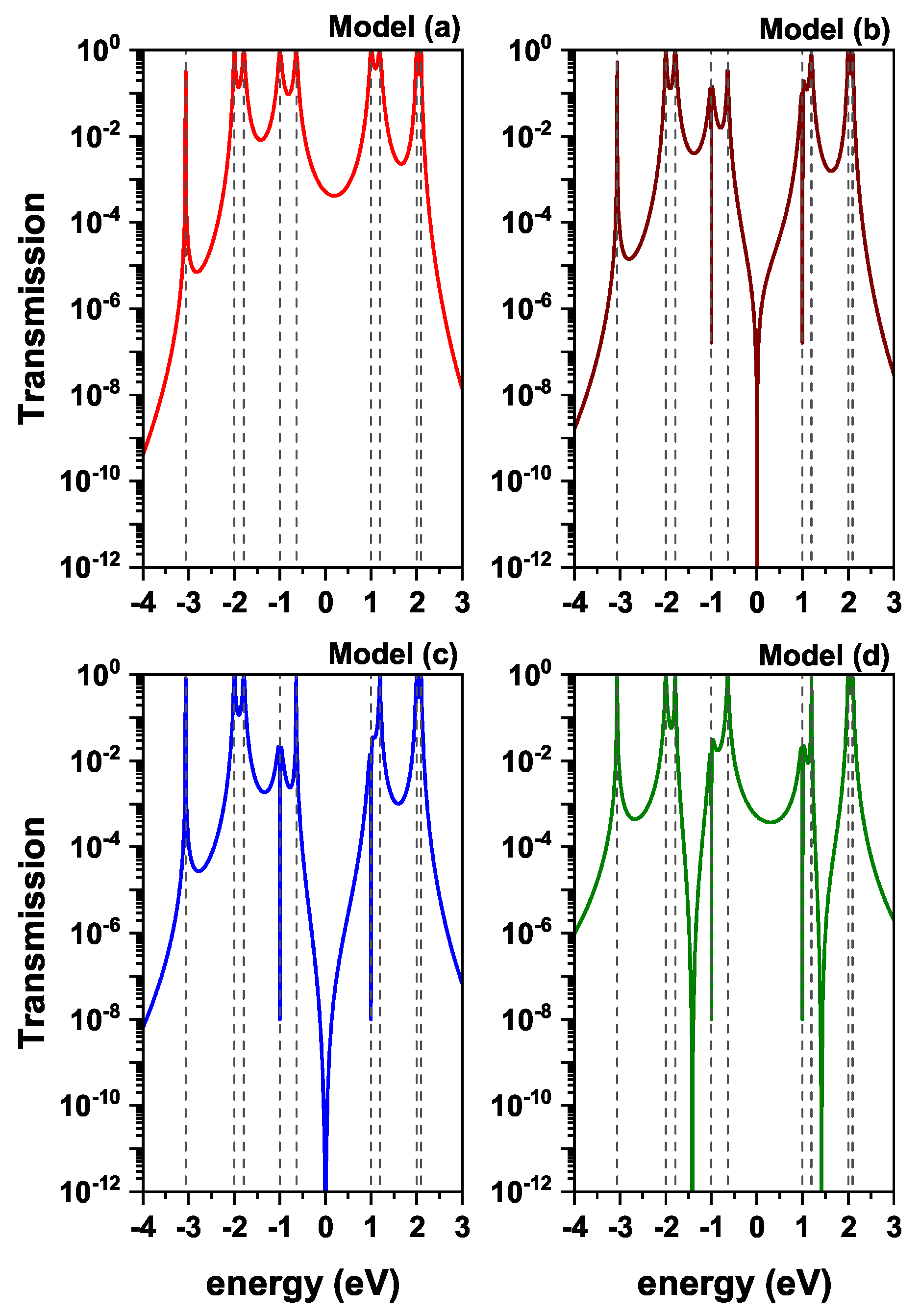
Figure 3 presents the transmission profile as a function of the incident electron energy, for a value of
eV (weak coupling).
The resonant peaks observed in the transmission plot are associated with the eigenvalues of the diphenyl ether molecule. On the other hand, 9 resonances are seen (degenerate and non-degenerate) in model (a), which are related to the 13 eigenvalues of the molecule (
eV); eigenvalues (
eV) are triple degenerate. When considering weak coupling,
eV, it implies that the electronic states of the electrodes have not mixed with those of the molecule. For this reason, there are quite defined peaks. Meanwhile, for all models, there are antiresonances with some eigenvalues of the molecule, associated with destructive quantum interference. This fact can be explained by means of Feynman integrals, where the electron can traverse all possible paths within the molecule and, depending on the paths chosen, can arrive at the drain electrode in phase (constructive interference) or out of phase (destructive interference). For model (a), the antiresonance is at
eV; for model (b), at
,
eV; for model (c), at
,
eV; and for model (d), at
,
eV,
eV. Furthermore, if these paths cancel in pairs, it leads to a node at the transmission probability at
[
42], as in the case of models (b) and (c).
4.1. Cofactor
Figure 4 shows the graphs of the cofactor comparison with the Green functions, as well as the effective coupling to the two sites of all models (a, b, c, and d), in the weak coupling regime (
eV). In model figure (a), there are two roots for the cofactor (
), but there are no crossings with the Green’s function (
), nor with the effective coupling (
) on the cofactor zeros. This agrees with what is shown in the transmission plot of
Figure 3 for model (a), in which we do not have destructive quantum interference. For models (b) and (c), there are three roots for
, and there are three crossings at these energy values, one for zero energy with
(model (b)), and
(model (c)), and two with energy −1 eV and 1 eV, with the
and
. Comparing these results with the graph of the transmission of
Figure 3 for models (b) and (c), it is expected that it presents the three destructive quantum interferences at 0, 1, and −1 eV. For model (d), we have a similar analysis, but this model presents four cofactor crossings, two with
, at energies of −1.4 and 1.4 eV, and two with
at −1 and 1 eV, and this agrees with what is shown in
Figure 3, where, for the d model, there are four destructive quantum interferences. This analysis allows us to verify the results obtained for the probability of transmission. The Green’s function
and the effective coupling
represent model (a), where the molecule is connected to sites 1 and 11. On the other hand,
and
correspond to model (b), in which the molecule is connected to the electrodes of sites 2 and 11. Regarding model (c),
and
are used when the molecule is connected to the electrodes of sites 2 and 10. Finally, for the system connected through sites 9 and 3 (model (d)),
and
are employed.
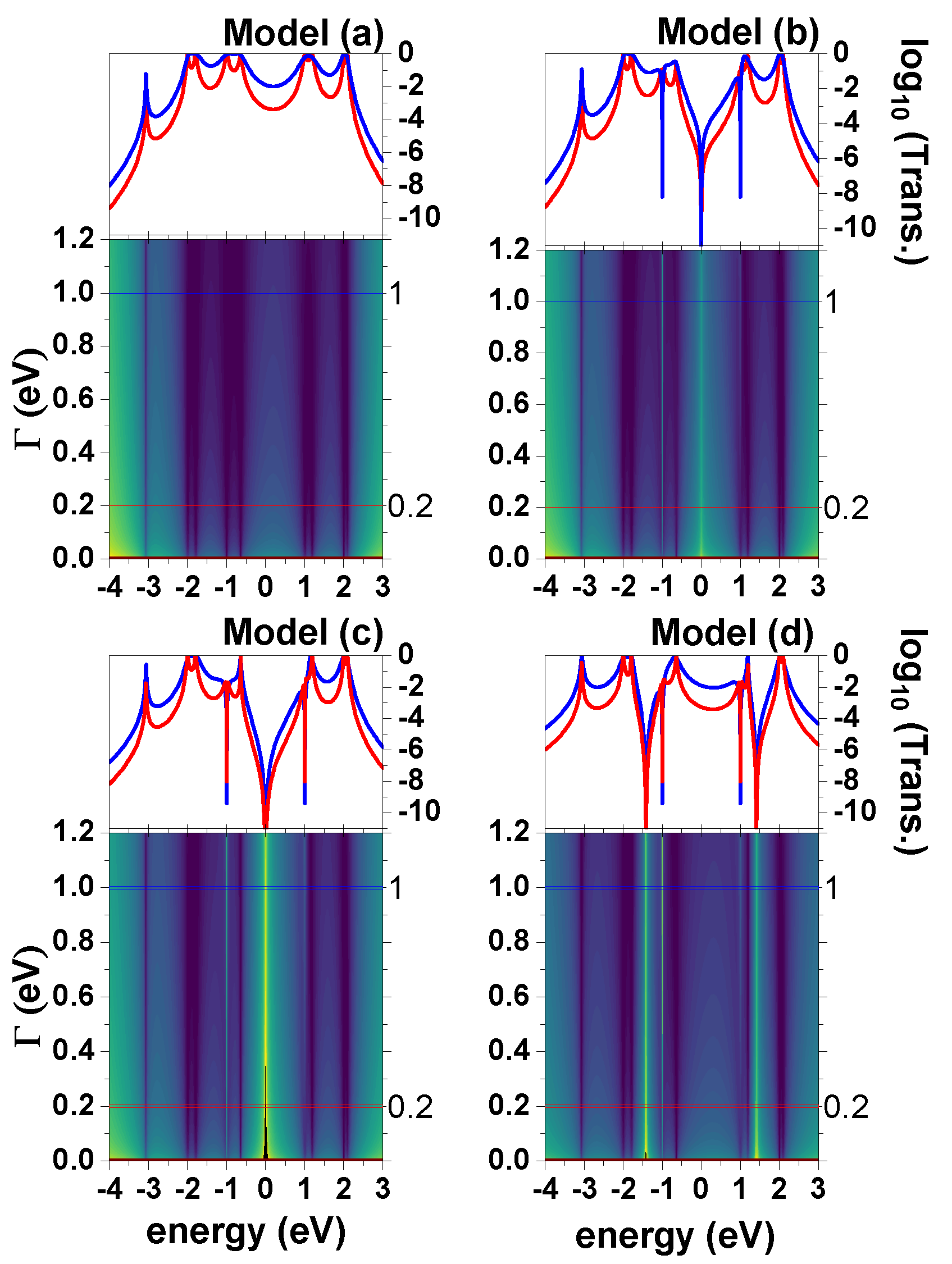
4.2. Transmission by Varying
Figure 5 shows a sweep in both
and the energy of the incident electron for the diphenyl-ether molecular system, for different coupling sites of the molecule and the metal electrodes.
At the bottom of the plots is a contour plot of the transmission probability logarithm, on the vertical axis is the sweep in , and on the horizontal axis is the sweep in the energy of the incident electron. In the upper part is the probability of the transmission logarithm as a function of the incident electron energy, for two different values of , one of weak coupling (0.2 eV), and another for strong coupling (1 eV). For the strong coupling, in all the models the loss of some peaks is observed in the transmission probability graph (upper graph). This is due to the peaks overlapping since the parameter is related to the peak at half maximum. This is due to the hybridization that exists between the delocalized states of the electrode and the localized states of the molecular system. For example, in model (a), for weak coupling (red graph) for energies of eV, we have two close peaks; something similar happens for energies of , 1, and 2 eV, but when there is strong coupling, these two peaks combine, forming one. This same analysis can be completed for models (b)–(d).
4.3. Current-Voltage Characteristics
Figure 5 shows the plot of current versus voltage with the Fermi equilibrium energy
set at 0 eV and a temperature of 300 K.
Figure 6 shows the plot of current versus voltage, the Fermi equilibrium energy
was set at 0 eV and the temperature at 300 K. It is noticed that in regions of constant current, where the system is far from the transmission resonances (see
Figure 3), and steep current regions where the transmission resonances are located. Furthermore, the current curves are antisymmetric with respect to zero volts. From
Figure 6, it can be seen that as the voltage increases, the gap between the electrodynamic potentials of the electrodes (left and right) becomes larger, and when the value of the electrodynamic potential coincides with a value characteristic of the molecule, there is a jump in the injection energy of the electron, until reaching a saturation value, which is when the molecule cannot store more electrons. Hence, the injection energy becomes constant and there will be no more energy jumps, no matter how much the voltage is increased. This saturation value is reached for a voltage of approximately 4 volts; at this point, a maximum current amplitude is obtained. Model (a) is the one with the greatest amplitude in the current, and is to be expected, since it presents the greatest area under the curve in the transmission graph, followed by model (b), model (c), and finally model (d). In the systems shown in the
I vs.
V graph, all present a value of 0 current in the voltage range of −1 volt and 1 volt, which indicates that the systems are not conductive, but it can be considered a semiconductor since they present this gap, which is due to the small transmission value seen in
Figure 3. This feature can be useful for designing an electronic switch.
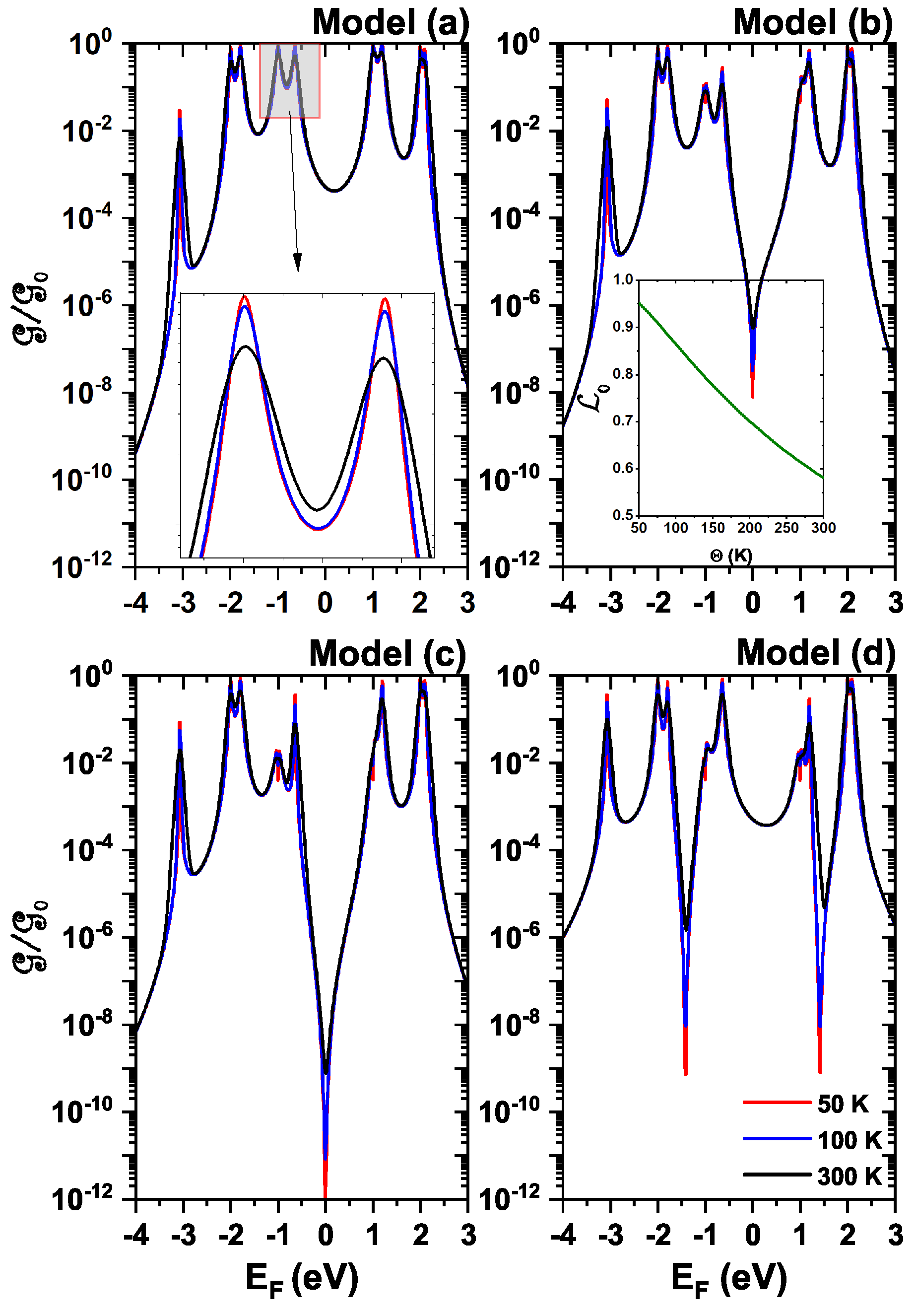
4.4. Electrical Conductance
Figure 7 shows the electrical conductance of a single diphenyl ether molecule, as a function of the Fermi energy, for models (a)–(d), for 3 different temperatures 50 K (red), 100 K (blue), and 300 K (black). The calculations are in the weak coupling regime (
eV); that is, the behavior of the molecule will be studied when the electrons pass through it. For all the models shown in the electrical conductance graph, it can be seen that it is proportional to the transmission probability (see
Figure 3 and Equation (
13)); that is, they share the same number of resonant peaks in the same energy positions. In all models, it can be noted that a forbidden band is generated, which is between −0.63 eV and 1 eV, due to destructive quantum interference between the localized states of the molecule and the delocalized states of the electrodes. At first glance, not many changes in conductance are noticeable for the different temperatures, but in the inset image of the model (a), it can be seen that the conductance decreases as the temperature increases. This decrease is a consequence of the derivative decrease, with respect to the energy, of the Fermi-Dirac distribution function in the integrand of the conductance Equation (
13), as the temperature increases (see the insert of
Figure 7 corresponding to model (b)).
The decrease in conductance is also related to scattering by molecular vibrations, the mechanism is not included in the present work. This occurs due to the fact that when the temperature increases, there is an increase in the electron’s kinetic energy since they move faster, thus increasing the amplitude of vibration of the atoms of the molecular system, thus behaving as a harmonic oscillator. This phenomenon causes the resistance to the passage of free electrons to increase since it is difficult to pass from one electrode to the other since the interference of the atoms with the trajectories of the valence electrons throughout the molecule increases.
4.5. Seebeck Coefficient
The thermoelectric properties are now shown, starting with the Seebeck coefficient, then the thermal conductance, and finally, the or figure of merit, taking into account the calculations of the previous physical quantities, to determine the efficiency of the models to convert thermal energy into electricity.
Figure 8 shows the Seebeck coefficient (
S) in a single molecule of diphenyl-ether, as a function of the Fermi energy, for models (a)–(d). The graph shows calculations for 3 different temperatures 50 K (red), 100 K (blue), and 300 K (black). The calculations are in a weak coupling regime (
eV). The
S is extracted from the results close to the HOMO−LUMO gap.
Table 1 shows parameters taken from
Figure 3,
Figure 7 and
Figure 8, for 300 K. It is observed that the Fermi energy is closer to the HOMO level, indicating a positive
S value for all models considered. Furthermore, it is verified that high
S values are coupled with low conductance values. The most notable result is that the
S is higher for models (b) and (c) where an antiresonance occurs at the Fermi energy, which coincides with the low HOMO conductance. It is important to note that the low temperature Seebeck coefficient results are very small due to the
(Equation (
12)), increasing magnitude as the temperature decreases (see the insert of
Figure 7 corresponding to model (b)).
4.6. Thermal Conductance
Now, we show the thermal conductance as a function of the Fermi energy, for a weak coupling of eV, and for different temperatures.
Figure 9 shows that thermal conductance increases as temperature increases, unlike electrical conductance. This is due to the fact that the increase in temperature, the integral of
and
, increases in value (see inset in the
Figure 9 corresponding to the model (d)). From Equation (15), the electrical conductance depends on the sum of
and the division of
by
, although the latter decreases with temperature, gain
. Another way of looking at thermal conductance is directly related to the increase in vibration of the atoms in the molecule, and these vibrations increase the average speed of the particles and therefore increase heat transfer, which is related to thermal conductance of the proposed models. The one with the greatest amplitude in thermal conductance is (a); this is related to the symmetry this model presents since the electrons leave one electrode to another. This is because when dividing the molecule, like the molecule travels, they recombine through the same number of sites, arriving in phase (see
Figure 3).
4.7. Figure of Merit ZT
Finally, we have in
Figure 10 the
, or figure of merit, shown as a function of the Fermi energy (
) to carry out the calculations of the
the results obtained for
G,
S, and
. Of the figures for the
, the model that presents the highest value is (c), which is consistent with the calculations obtained for the Seebeck Coefficient (see
Table 1), since it is the model that presents the highest
S, and the higher the
S, the higher the value of
. This is due to the fact that model (c) presents an interference in the graph of the transmission probability around the Fermi level, and this is more pronounced than in model (b), which is the model with the second highest
.
The model with the smallest
is presented by (a); therefore, it is with the smallest
S, which is the most symmetrical model, and does not present interference in its transmission. For all the models shown, the
. These values indicate that the systems studied can present a good efficiency in the conversion of thermoelectric energy, which is slightly higher than the
ZT value of the commercially available inorganic semiconductor, bismuth telluride (Bi
Te
), which has a
ZT of 1 [
43]. Lastly, it is important to mention that the higher the temperature, the higher the efficiency.

 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}