
Generation of Viral Particles with Brain Cell-Specific Tropism by Pseudotyping HIV-1 with the Zika Virus E Protein
 , ,
, ,
Abstract
:1. Introduction
2. Experimental Design
2.1. General Prerequisites
2.1.1. Plasmid DNA
2.1.2. Cells for Viral Packaging
2.1.3. Cells for Transduction
2.2. Equipment
- Heraeus microcentrifuge, Biofuge pico #11332 (Thermo Scientific, Dreieich, Germany).
- Biological Safety Cabinet, Sterilgard III Advance Class II (Baker Company, Utrecht, The Netherlands).
- Fluorescence microscope, EVOS M7000 (Thermo Fisher Scientific, Dreieich, Germany).
- CO2 Incubator CB 150 (BINDER, Tuttlingen, Germany).
- UV-VIS Spectrophotometer, ShimadzuUV160A, with supermicro cell holder for measurement of 100 µL samples. Using a supermicro black cell of fused silica, 10 mm path, #206-14334. (Shimadzu, Duisburg, Germany).
- Micropipettes, Eppendorf Research® plus, 0.5-10 µL, 5-100 µL (Eppendorf, Hamburg, Germany).
2.3. Materials
- Pipette tips, filter tip, transparent, Biosphere® plus, low retention, 10 µL, 100 µL, 200 µL (Sarstedt, Nümbrecht, Germany).
- Reaction tubes, SafeSeal reaction tube, 1.5 mL, PP #72.706Sarstedt AG & Co. KG (Sarstedt, Nümbrecht, Germany).
- Cell culture plates, 24-well, standard surface, flat base #83.3922 (Sarstedt, Nümbrecht, Germany).
- Cell culture plates, 96-well, Cell+ surface, flat base, #83.3924.300 (Sarstedt, Nümbrecht, Germany).
- Cell culture pipettes, 5 mL, 10 mL, 25 mL (Sarstedt, Nümbrecht, Germany).
2.4. Reagents
- Cell culture medium, DMEM, w: 4.5 g/L glucose, w: stable glutamine, w: sodium pyruvate, w: 3.7 g/L NaHCO3, #P04-4515, (Pan-Biotech, Aidenbach, Germany).
- Medium supplement, FBS Good, EU-approved regions, filtrated bovine serum, 0.2 µm sterile filtered PA0-37500 (Pan-Biotech, Aidenbach, Germany).
- Human cerebrospinal fluid (hCSF) (Asklepios Heidberg Nord, Hamburg, Germany). Use of hCSF was approved by the Ethical Commission of the Hamburg Medical Chamber (Hamburg, Germany) with registration number PV6041.
- Transfection reagent and dilution buffer, ScreenFect®A (SFA) (ScreenFect, Eggenstein-Leopoldshafen, Germany).
- Transfection-grade plasmid DNA isolation, NucleoBond Xtra Midi kit, #740410.100 (Macherey-Nagel, Düren, Germany).
2.5. Plasmids
- pNLgfpAM, Nikolas Friedrich, Alexandra Trkola, Institute of Medical Virology, University of Zürich, Switzerland.
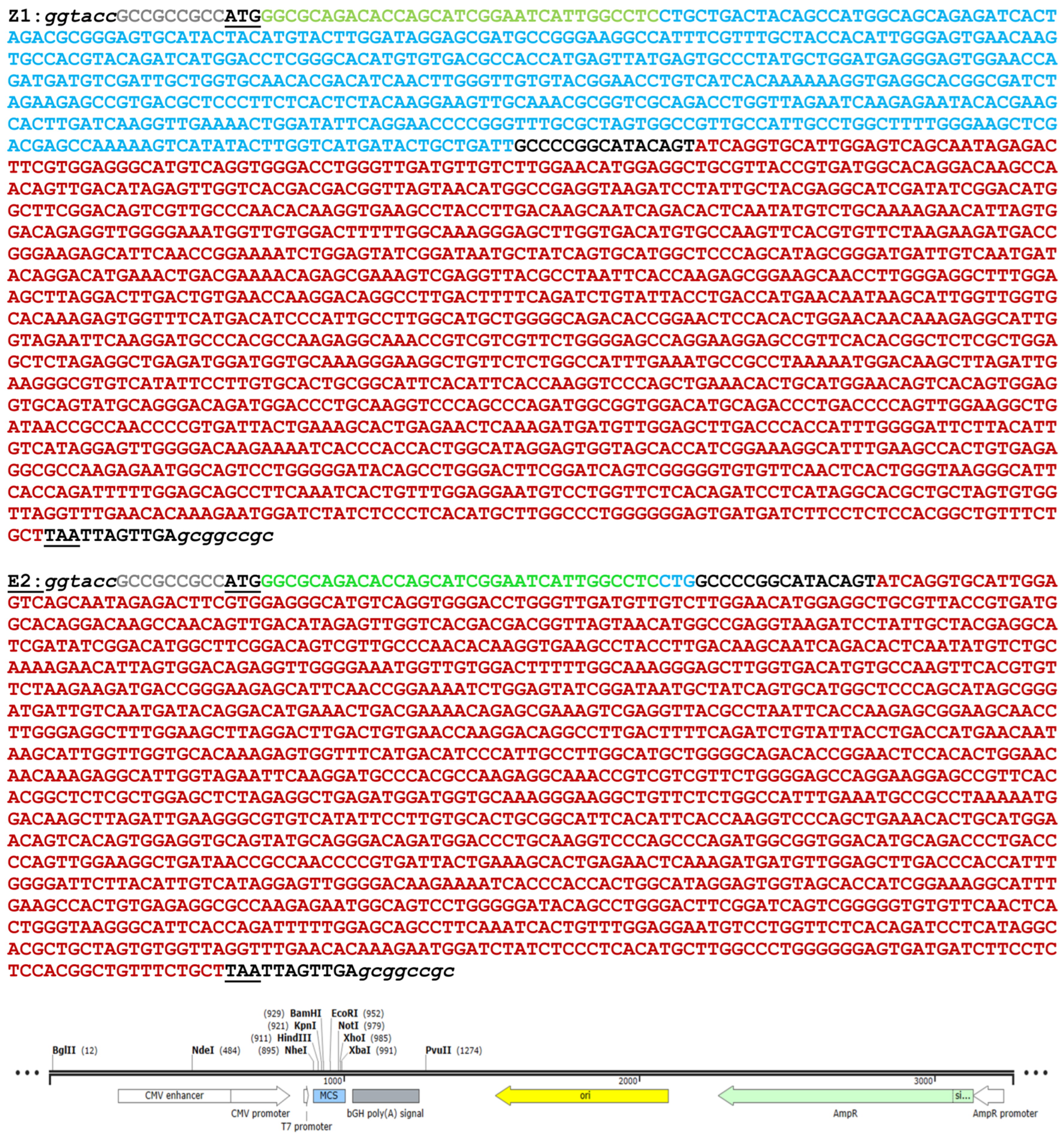
- pME-Z1, LG Schreiber, Bernhard Nocht Institute for Tropical Medicine, Hamburg, Germany [8].
- pME-E2, LG Schreiber, Bernhard Nocht Institute for Tropical Medicine, Hamburg, Germany [16].
- pCMV-VSV-G, addgene plasmid #8454, (addgene, Watertown, MA, USA).
2.6. Cells
- COS-1, Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, no.: ACC 63.
- AKH-16, primary cell culture made from tissue samples of a GBM tumor. LG Schreiber, Bernhard Nocht Institute for Tropical Medicine, Hamburg, Germany. Use of human tumor samples was approved by the Ethical Commission of the Hamburg Medical Chamber (Hamburg, Germany) with registration number PV6041.
3. Procedure
3.1. Production of Pseudotyped HIV-1 Particles
3.1.1. Day One
- Add 1000 µL of a COS-1 cell suspension (see note in Section 6.2) in DMEM/10% FBS to a well of a 24-well plate. The cell suspension should be adjusted so that the cells do not become too dense on day 3, as there is a risk that they will otherwise detach. If the density is too low, the transfection will result in low pseudotype production. Therefore, it is recommended to perform preliminary experiments to investigate the growth of COS-1 cells under the respective laboratory conditions to get the best possible transfection results. However, the cells should have a confluence of 70–80% on the day of transfection.
3.1.2. Day Two
- Replace the old medium with 1000 µL of fresh DMEM/10% FBS medium.
- For the transfection procedure, the DNA and SFA reagent preparations are each mixed in a separate 1.5 mL reaction tube following the recipe shown in Table 1. The plasmid DNAs are all adjusted to a concentration of 1 mg/mL in H2O. Because of the cell toxicity of SFA, the amount of SFA reagents should not be increased to minimize the risk of COS-1 cell detachment.
- Mixture 2 is added to mixture 1 in 15–20 µL increments and mixed each time by pipetting up and down (5 × 20 µL pipette strokes). Mixture 2 must be slowly transferred into mixture 1 in small steps and mixed well to prevent precipitation of the DNA. The DNA/SFA mixture will be incubated for 20 min at room temperature. After incubation, carefully add the DNA/SFA mixture onto the 1000 µL of the COS-1 cell culture medium. Do not mix the DNA/SFA mixture with the cell culture medium by pipetting up and down. Gently agitate the 24-well plates back and forth 2–3 times and transfer the plates carefully into the CO2 incubator. Incubate the 24-well plates for 24 h at 37 °C and 5% CO2.
3.1.3. Day Three
- The medium also containing the DNA/SFA mixture is carefully replaced with 1000 µL of fresh DMEM/10% FBS and the cells are incubated for another 48 h at 37 °C and 5% CO2. In this step, it is important to remove any free plasmid DNA that may be present in the cell culture supernatant. This is particularly important when using plasmids containing firefly luciferase.
- For the pseudotype experiments, the 96-well plates should now be prepared. Therefore, 200 µL of an AKH-16 cell suspension grown in CSF-DF medium (50% hCSF, 45% DMEM, 5% FBS) was added to a well of a 96-well plate. CSF-DF, a 1:1 mixture of DMEM/10% FBS and hCSF has been shown to be an excellent medium for the growth of cell cultures made from glioblastoma tissue samples that maintain their heterogeneity over a long period of time.
3.2. Transduction of GBM Tumor Cells
3.2.1. Day Five
- To verify successful pNLgfpAM transfection and pseudotype production, examine the wells using the EVOS M7000 fluorescence microscope (GFP channel, see note in Section 6.3).
- To collect pseudotype particles, carefully remove the cell culture medium from the transfected cultures without touching the bottom of the well with the pipette tip to avoid picking up of COS-1 cells. The cell culture supernatant is collected into a 1.5 mL reaction tube and centrifuged at maximum speed (13,000 rpm) for 2 min at RT. After centrifugation, carefully transfer the medium to a new reaction tube by touching the surface of the medium with the pipette tip and, finally, leaving 50–100 µL of the liquid at the bottom of the tube. Avoid picking up liquid from the bottom of the tube. The supernatant is now centrifuged for a second time (13,000 rpm, 2 min, RT) and the supernatant is again transferred into a new reaction tube. This procedure yields 800–900 µL of pseudotype containing cell culture medium for every transfected 24-well.
- For optimal transduction, the confluence of the primary AKH-16 cell cultures should be around 70–80% on the day the pseudotype is added. Depending on the pseudotype yield, the supernatant can be used directly or diluted in medium. As a rule, 100 µL of a pseudotype-containing medium is added to the tumor cells and the 96-well plates are incubated at 37 °C and 5% CO2 for 24 h.
3.2.2. Day Six
- The next day, the CSF-DF medium is changed, and the plates are incubated for another 48 h at 37 °C and 5% CO2. This step is important especially when firefly luciferase is used as a reporter gene. In this case, it is important that the cell culture supernatants are free of any luciferase activity. Therefore, several washing steps may be required 24 h after transfection to remove luciferase activity from the cell culture supernatant while leaving the cell layer intact.
3.2.3. Day Eight
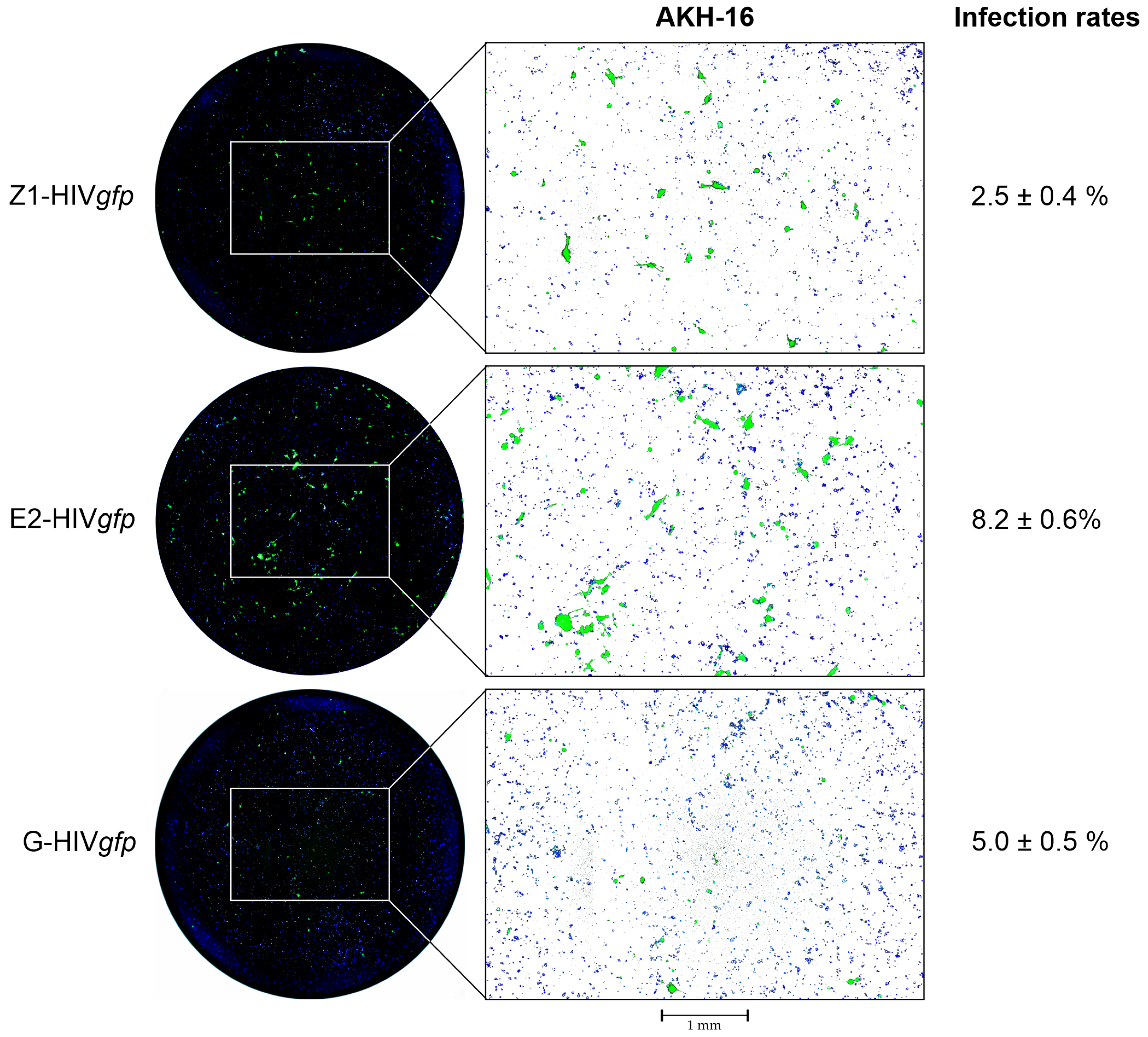
- For the detection of pseudotype transduced cells using GFP as a reporter, the plates including the medium are examined using the EVOS M7000 fluorescence microscope using the GFP (470/525 nm) and DAPI (357/447 nm) channels (excitation/emission). The 96-well area is scanned at 4× magnification and the pictures are automatically stitched together by the EVOS M7000 imaging software. The successful transduction of AKH-16 cell cultures with Z1-HIVgfp, E2-HIVgfp or G-HIVgfp pseudotypes is shown in Figure 4.
4. Discussion
5. Conclusions
6. Notes
6.1. Making Plasmid Stocks for Transfection
6.2. Making Plates for Transfection and Transduction
6.3. Checking Transfection Efficiency
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Wolff, J.H.; Mikkelsen, J.G. Delivering Genes with Human Immunodeficiency Virus-Derived Vehicles: Still State-of-the-Art after 25 Years. J. Biomed. Sci. 2022, 29, 79. [Google Scholar] [CrossRef] [PubMed]
- Cronin, J.; Zhang, X.-Y.; Reiser, J. Altering the Tropism of Lentiviral Vectors through Pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with Oncolytic Viruses: Progress and Challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic Virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika Virus Has Oncolytic Activity against Glioblastoma Stem Cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef]
- Rius-Rocabert, S.; García-Romero, N.; García, A.; Ayuso-Sacido, A.; Nistal-Villan, E. Oncolytic Virotherapy in Glioma Tumors. Int. J. Mol. Sci. 2020, 21, 7604. [Google Scholar] [CrossRef] [PubMed]
- Foreman, P.M.; Friedman, G.K.; Cassady, K.A.; Markert, J.M. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurotherapeutics 2017, 14, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, M.; Kadlubowska, P.; Hoffmann, D.; Schwalbe, B.; Auerswald, H.; Schreiber, M. Zikavirus Pr ME Envelope Pseudotyped Human Immunodeficiency Virus Type-1 as a Novel Tool for Glioblastoma-Directed Virotherapy. Cancers 2020, 12, 1000. [Google Scholar] [CrossRef]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A.; et al. Axl and Growth Arrest-Specific Gene 6 Are Frequently Overexpressed in Human Gliomas and Predict Poor Prognosis in Patients with Glioblastoma Multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef]
- Zwernik, S.D.; Adams, B.H.; Raymond, D.A.; Warner, C.M.; Kassam, A.B.; Rovin, R.A.; Akhtar, P. AXL Receptor Is Required for Zika Virus Strain MR-766 Infection in Human Glioblastoma Cell Lines. Mol. Ther. Oncolytics 2021, 23, 447–457. [Google Scholar] [CrossRef]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.-M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Min, K.-I.; Lee, J.; Kang, S.-H.; Jeon, W.; Nam, J.H.; Ju, Y.R.; Kim, Y.B. The prM-Independent Packaging of Pseudotyped Japanese Encephalitis Virus. Virol. J. 2009, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, R.; Yuan, L.; Tian, G.; Huang, X.; Wen, Y.; Ma, X.; Huang, Y.; Yan, Q.; Zhao, Q.; et al. Introducing a Cleavable Signal Peptide Enhances the Packaging Efficiency of Lentiviral Vectors Pseudotyped with Japanese Encephalitis Virus Envelope Proteins. Virus Res. 2017, 229, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mao, Y.; Li, Q.; Qiu, Z.; Li, J.; Li, X.; Liang, W.; Xu, M.; Li, A.; Cai, X.; et al. Efficient Gene Transfer to Kidney Using a Lentiviral Vector Pseudotyped with Zika Virus Envelope Glycoprotein. Hum. Gene Ther. 2022, 33, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Pöhlking, C.; Beier, S.; Formanski, J.P.; Friese, M.; Schreiber, M.; Schwalbe, B. Isolation of Cells from Glioblastoma Multiforme Grade 4 Tumors for Infection with Zika Virus prME and ME Pseudotyped HIV-1. Int. J. Mol. Sci. 2023, 24, 4467. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, V.; Ngo, H.D.; Formanski, J.P.; Jonas, J.S.; Pöhlking, C.; Schwalbe, B.; Schreiber, M. Development of Zika Virus E Variants for Pseudotyping Retroviral Vectors Targeting Glioblastoma Cells. Int. J. Mol. Sci. 2023, 24, 14487. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.-J.J.; Hunt, A.R.; Holmes, D.A.; Springfield, T.; Chiueh, T.-S.; Roehrig, J.T.; Gubler, D.J. Enhancing Biosynthesis and Secretion of Premembrane and Envelope Proteins by the Chimeric Plasmid of Dengue Virus Type 2 and Japanese Encephalitis Virus. Virology 2003, 306, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Hassn Mesrati, M.; Behrooz, A.B.; Abuhamad, A.Y.; Syahir, A. Understanding Glioblastoma Biomarkers: Knocking a Mountain with a Hammer. Cells 2020, 9, 1236. [Google Scholar] [CrossRef]
- Wilson, C.B.; Barker, M. Cerebrospinal Fluid as a Culture Medium for Human Brain Tumors. Neurology 1966, 16, 1064. [Google Scholar] [CrossRef]
- Hu, H.P.; Hsieh, S.C.; King, C.C.; Wang, W.K. Characterization of Retrovirus-Based Reporter Viruses Pseudotyped with the Precursor Membrane and Envelope Glycoproteins of Four Serotypes of Dengue Viruses. Virology 2007, 368, 376–387. [Google Scholar] [CrossRef]
- Ruiz-Jiménez, F.; Pérez-Olais, J.H.; Raymond, C.; King, B.J.; McClure, C.P.; Urbanowicz, R.A.; Ball, J.K. Challenges on the Development of a Pseudotyping Assay for Zika Glycoproteins. J. Med. Microbiol. 2021, 70, 001413. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, D.O.; Landau, N.R. Human Immunodeficiency Virus Type 1 Viral Protein R (Vpr) Arrests Cells in the G2 Phase of the Cell Cycle by Inhibiting P34cdc2 Activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [CrossRef] [PubMed]
- Stiasny, K.; Kiermayr, S.; Bernhart, A.; Heinz, F.X. The Membrane-Proximal “Stem” Region Increases the Stability of the Flavivirus E Protein Postfusion Trimer and Modulates Its Structure. J. Virol. 2013, 87, 9933. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. An Analysis of 5’-Noncoding Sequences from 699 Vertebrate Messenger RNAs. Nucleic Acids Res. 1987, 15, 8125–8148. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixture 1 (DNA) | Mixture 2 (SFA) | ||
|---|---|---|---|
| Envelope plasmid 1 | 9.25 µL | SFA reagents | 15.0 µL |
| pNLgfpAM | 2.00 µL | SFA dilution buffer | 34.5 µL |
| SFA dilution buffer | 30.00 µL | ||
| Ʃ 41.25 µL | Ʃ 49.5 µL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngo, H.D.; Formanski, J.P.; Grunwald, V.; Schwalbe, B.; Schreiber, M. Generation of Viral Particles with Brain Cell-Specific Tropism by Pseudotyping HIV-1 with the Zika Virus E Protein. Methods Protoc. 2024, 7, 3. https://doi.org/10.3390/mps7010003
Ngo HD, Formanski JP, Grunwald V, Schwalbe B, Schreiber M. Generation of Viral Particles with Brain Cell-Specific Tropism by Pseudotyping HIV-1 with the Zika Virus E Protein. Methods and Protocols. 2024; 7(1):3. https://doi.org/10.3390/mps7010003
Chicago/Turabian StyleNgo, Hai Dang, Jan Patrick Formanski, Vivien Grunwald, Birco Schwalbe, and Michael Schreiber. 2024. "Generation of Viral Particles with Brain Cell-Specific Tropism by Pseudotyping HIV-1 with the Zika Virus E Protein" Methods and Protocols 7, no. 1: 3. https://doi.org/10.3390/mps7010003