
Simple Acid Digestion Procedure for the Determination of Total Mercury in Plankton by Cold Vapor Atomic Fluorescence Spectroscopy


Abstract
:1. Introduction
2. Material and Methods
2.1. Materials and Reagents
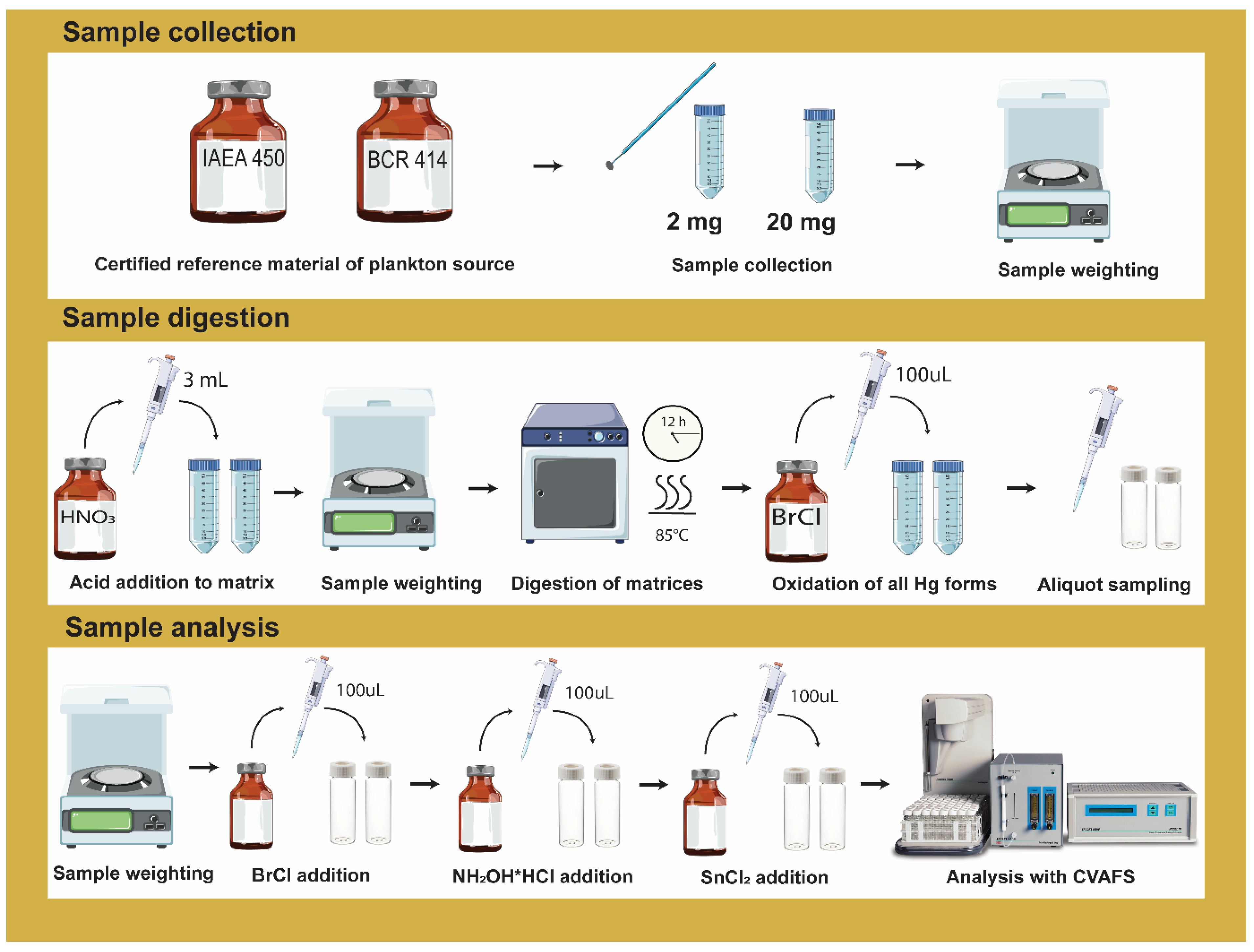
2.2. Sample Preparation and Digestion
2.3. Total Hg Analysis
2.4. Analytical Quality Assurance and Method Validation Parameters
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, Y.; Shi, Z.M.; Long, Z.; Wu, P.; Zheng, C.B.; Hou, X.D. Determination and speciation of mercury in environmental and biological samples by analytical atomic spectrometry. Microchem. J. 2012, 103, 1–14. [Google Scholar] [CrossRef]
- Harding, G.; Dalziel, J.; Vass, P. Bioaccumulation of methylmercury within the marine food web of the outer Bay of Fundy, Gulf of Maine. PLoS ONE 2018, 13, e0197220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.S.; Fisher, N.S. Bioaccumulation of methylmercury in a marine diatom and the influence of dissolved organic matter. Mar. Chem. 2017, 197, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Lehnherr, I. Methylmercury biogeochemistry: A review with special reference to Arctic aquatic ecosystems. Environ. Rev. 2014, 22, 229–243. [Google Scholar] [CrossRef]
- Dranguet, P.; Fluck, R.; Regier, N.; Cosio, C.; Le Faucheur, S.; Slaveykova, V.I. Towards Mechanistic Understanding of Mercury Availability and Toxicity to Aquatic Primary Producers. CHIMIA Int. J. Chem. 2014, 68, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Chasar, L.C.; Scudder, B.C.; Stewart, A.R.; Bell, A.H.; Aiken, G.R. Mercury cycling in stream ecosystems. 3. Trophic dynamics and methylmercury bioaccumulation. Environ. Sci. Technol. 2009, 43, 2733–2739. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, T.; Ting, Y.-P. Selection of wet digestion methods for metal quantification in hazardous solid wastes. J. Environ. Chem. Eng. 2015, 3, 1459–1467. [Google Scholar] [CrossRef]
- Cizdziel, J.V.; Tolbert, C.; Brown, G. Direct analysis of environmental and biological samples for total mercury with comparison of sequential atomic absorption and fluorescence measurements from a single combustion event. Spectrochim. Acta Part B At. Spectrosc. 2010, 65, 176–180. [Google Scholar] [CrossRef]
- Bussan, D.D.; Sessums, R.F.; Cizdziel, J.V. Direct mercury analysis in environmental solids by ICP-MS with on-line sample ashing and mercury pre-concentration using a direct mercury analyzer. J. Anal. At. Spectrom. 2015, 30, 1668–1672. [Google Scholar] [CrossRef]
- Suvarapu, L.N.; Baek, S.O. Recent Studies on the Speciation and Determination of Mercury in Different Environmental Matrices Using Various Analytical Techniques. Int. J. Anal. Chem. 2017, 2017, 3624015. [Google Scholar] [CrossRef] [Green Version]
- Spanu, D.; Butti, L.; Boldrocchi, G.; Bettinetti, R.; Monticelli, D. High-throughput, Multi-batch System for the Efficient Microwave Digestion of Biological Samples. Anal. Sci. 2020, 36, 889–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voegborlo, R.B.; Adimado, A.A. A simple classical wet digestion technique for the determination of total mercury in fish tissue by cold-vapour atomic absorption spectrometry in a low technology environment. Food Chem. 2010, 123, 936–940. [Google Scholar] [CrossRef]
- Lomonte, C.; Gregory, D.; Baker, A.J.M.; Kolev, S.D. Comparative study of hotplate wet digestion methods for the determination of mercury in biosolids. Chemosphere 2008, 72, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, E.; Mohammed, T.; Mohammed, A. Optimization of an acid digestion procedure for the determination of Hg, As, Sb, Pb and Cd in fish muscle tissue. MethodsX 2017, 4, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Frentiu, T.; Mihaltan, A.I.; Senila, M.; Darvasi, E.; Ponta, M.; Frentiu, M.; Pintican, B.P. New method for mercury determination in microwave digested soil samples based on cold vapor capacitively coupled plasma microtorch optical emission spectrometry: Comparison with atomic fluorescence spectrometry. Microchem. J. 2013, 110, 545–552. [Google Scholar] [CrossRef]
- Taylor, V.F.; Jackson, B.P.; Chen, C.Y. Mercury speciation and total trace element determination of low-biomass biological samples. Anal. Bioanal. Chem. 2008, 392, 1283–1290. [Google Scholar] [CrossRef] [Green Version]
- Yanez-Jacome, G.S.; Romero-Estevez, D.; Navarrete, H.; Simbana-Farinango, K.; Velez-Terreros, P.Y. Optimization of a Digestion Method to Determine Total Mercury in Fish Tissue by Cold Vapor Atomic Fluorescence Spectrophotometry. Methods Protoc. 2020, 3, 45. [Google Scholar] [CrossRef]
- Navarro, P.; Raposo, J.C.; Arana, G.; Etxebarria, N. Optimisation of microwave assisted digestion of sediments and determination of Sn and Hg. Anal. Chim. Acta 2006, 566, 37–44. [Google Scholar] [CrossRef]
- Lima, A.F.; Da Costa, M.C.; Ferreira, D.C.; Richter, E.M.; Munoz, R.A.A. Fast ultrasound-assisted treatment of inorganic fertilizers for mercury determination by atomic absorption spectrometry and microwave-induced plasma spectrometry with the aid of the cold-vapor technique. Microchem. J. 2015, 118, 40–44. [Google Scholar] [CrossRef]
- Han, F.X.X.; Patterson, W.D.; Xia, Y.J.; Sridhar, B.B.; Su, Y. Rapid determination of mercury in plant and soil samples using inductively coupled plasma atomic emission spectroscopy, a comparative study. Water Air Soil Pollut. 2006, 170, 161–171. [Google Scholar] [CrossRef]
- Arslan, Z.; Ertas, N.; Tyson, J.F.; Uden, P.C.; Denoyer, E.R. Determination of trace elements in marine plankton by inductively coupled plasma mass spectrometry (ICP-MS). Fresenius J. Anal. Chem. 2000, 366, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EPA. Method 1631: Mercury in Water by Oxidation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectrometry; OW, EPA-821-R-02-019; EPA: Washington, DC, USA, 2002.
- EPA. Appendix to Method 1631 Total Mercury in Tissue, Sludge, Sediment, and Soil by Acid Digestion and BrCI Oxidation; EPA: Washington, DC, USA, 2001.
- Cardellicchio, N.; Di Leo, A.; Giandomenico, S.; Santoro, S. Optimization of microwave digestion for mercury determination in marine biological samples by cold vapour atomic absorption spectrometry. Ann. Chim. 2006, 96, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Bravo, A.G.; Kothawala, D.N.; Attermeyer, K.; Tessier, E.; Bodmer, P.; Amouroux, D. Cleaning and sampling protocol for analysis of mercury and dissolved organic matter in freshwater systems. MethodsX 2018, 5, 1017–1026. [Google Scholar] [CrossRef]
- Thompson, M.; Ellison, S.L.R.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis—(IUPAC technical report). Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- Guideline, I.H.T. Validation of analytical procedures: Text and methodology Q2 (R1). In Proceedings of the International Conference on Harmonization, San Diego, CA, USA, 2 June 2014. [Google Scholar]
- Barwick, V. Guide to Quality in Analytical Chemistry an Aid to Accreditation, 3rd ed.; Eurachem: Teddington, UK, 2016. [Google Scholar]
- Magnusson, B.; Örnemar, U. The Fitness for Purpose of Analytical Methods. In Magnusson, U. Örnemark, Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; Eurachem: Teddington, UK, 2014. [Google Scholar]
- Ellison, S.L.; Barwick, V.J.; Farrant, T.J.D. Practical Statistics for the Analytical Scientist: A Bench Guide; Royal Society of Chemistry: London, UK, 2009. [Google Scholar]
- Elezz, A.A.; Mustafa Hassan, H.; Abdulla Alsaadi, H.; Easa, A.; Al-Meer, S.; Elsaid, K.; Ghouri, Z.K.; Abdala, A. Validation of total mercury in marine sediment and biological samples, using cold vapour atomic absorption spectrometry. Methods Protoc. 2018, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Pickhardt, P.C.; Fisher, N.S. Accumulation of inorganic and methylmercury by freshwater phytoplankton in two contrasting water bodies. Environ. Sci. Technol. 2007, 41, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, C.R.; Finiguerra, M.B.; Weller, R.L.; Fitzgerald, W.F. Methylmercury accumulation in plankton on the continental margin of the northwest Atlantic Ocean. Environ. Sci. Technol. 2013, 47, 3671–3677. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
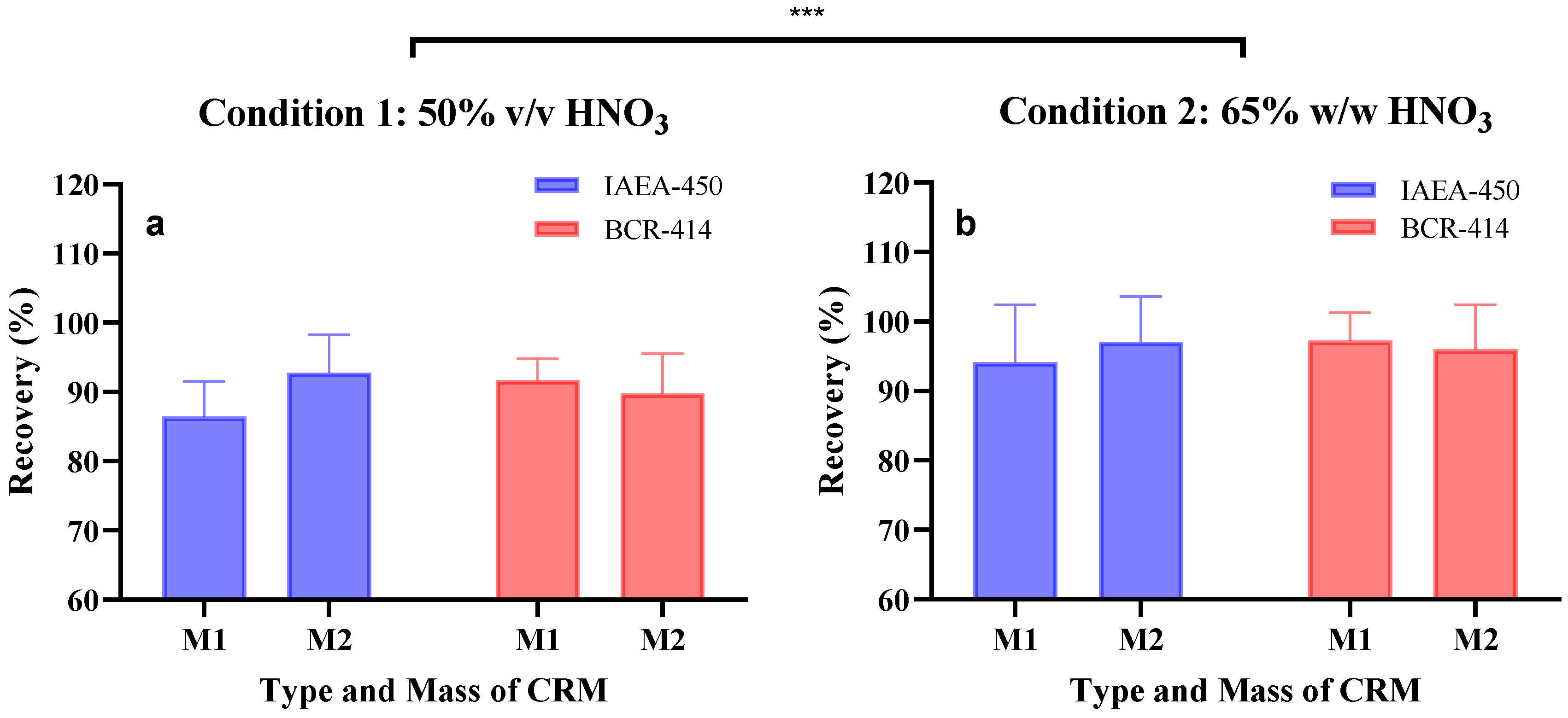
| Condition | CRM | Weight CRM (g) | Recovery (%) | BIAS (%) | Repeatability | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Mean | SDpooled | RSDpooled | Sr | |||||
| 1. | IAEA-450 | M1 0.0031 | ± | 0.0008 | 86.4% | 5.9% | 6.8% | −13.6% | 5.9% |
| M2 0.0195 | ± | 0.0010 | 92.8% | 6.0% | 6.4% | −7.3% | 11.9% | ||
| BCR-414 | M1 0.0028 | ± | 0.0008 | 91.7% | 3.4% | 3.7% | −8.5% | 4.8% | |
| M2 0.0216 | ± | 0.0034 | 89.8% | 4.8% | 5.4% | −10.5% | 4.8% | ||
| 2. | IAEA-450 | M1 0.0026 | ± | 0.0002 | 94.1% | 7.6% | 8.1% | −6.2% | 7.9% |
| M2 0.0231 | ± | 0.0042 | 97.1% | 7.5% | 7.7% | −3.2% | 7.7% | ||
| BCR-414 | M1 0.0041 | ± | 0.0008 | 97.2% | 4.6% | 4.7% | −2.9% | 4.6% | |
| M2 0.0221 | ± | 0.0048 | 96.0% | 6.9% | 7.2% | −4.5% | 6.9% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.P.; Mehmeti, L.; Slaveykova, V.I. Simple Acid Digestion Procedure for the Determination of Total Mercury in Plankton by Cold Vapor Atomic Fluorescence Spectroscopy. Methods Protoc. 2022, 5, 29. https://doi.org/10.3390/mps5020029
Santos JP, Mehmeti L, Slaveykova VI. Simple Acid Digestion Procedure for the Determination of Total Mercury in Plankton by Cold Vapor Atomic Fluorescence Spectroscopy. Methods and Protocols. 2022; 5(2):29. https://doi.org/10.3390/mps5020029
Chicago/Turabian StyleSantos, João Pereira, Lirie Mehmeti, and Vera I. Slaveykova. 2022. "Simple Acid Digestion Procedure for the Determination of Total Mercury in Plankton by Cold Vapor Atomic Fluorescence Spectroscopy" Methods and Protocols 5, no. 2: 29. https://doi.org/10.3390/mps5020029