An Optimised Step-by-Step Protocol for Measuring Relative Telomere Length

Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
2.1.1. DNA Isolation
- QIAamp™ Blood Midi kit (Qiagen®, Hilden, Germany; Cat. no.: 51185) or any other preferred/tested kit. The Qiagen® kit contains QIAamp Midi Spin Columns, Collection Tubes (15 ml), Buffer AL, Buffer AW1 (concentrate), Buffer AW2 (concentrate), Buffer AE and QIAGEN® Protease.
- Molecular biology grade ethanol (100% pure) (Sigma-Aldrich, St. Louis, MO, USA; Cat. no.: E7023)
2.1.2. Telomere and Single Copy Gene Quantitation by qPCR
- Fast SYBR Green master-mix (ThermoFisher Scientific, Waltham, MA, USA; Cat. no.: 4385617)
- A fresh aliquot of nuclease-free water (Qiagen, Hilden, Germany; Cat. no.: 129117)
- Primers for telomere as detailed in [23] (Integrated DNA Technologies, Coralville, IA, USA; Cat. no.: CUSTOM OLIGOS)Telomere (A): CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTTTelomere (B): GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT
- Primers for human β-globin as detailed in [24] (Integrated DNA Technologies, Coralville, IA, USA; Cat. no.: CUSTOM OLIGOS). The human β-globin gene is used as single-copy gene in our protocol.hbg1: GCTTCTGACACAACTGTGTTCACTAGChbg2: CACCAACTTCATCCACGTTCACC
2.1.3. Consumables
- 1.7 mL Eppendorf tubes (Axygen Scientific, Union City, SF, USA; Cat. no.: MCT-175-C)
- P10, P20, P200 and P1000 Filtered pipette tips (Axygen Scientific, Union City, SF, USA; Cat. no.: TF-300-L-R-S, TF-100-L-R-S, TF-200-L-R-S, and TF-1000-L-R-S, respectively)
- 15 mL conical tubes (Sigma-Aldrich, St. Louis, MO, USA; Cat. no.: CLS430791)
- MicroAmp™ Fast Optical 96-well PCR plates (0.1 mL volume of each well) (ThermoFisher Scientific, Waltham, MA, USA; Cat. no.: 4366932)
- MicroAmp™ Optical Adhesive Film for the PCR plates (ThermoFisher Scientific, Waltham, MA, USA; Cat. no.: 4311971)
2.2. Equipment
- Water bath that can heat up to 70 °C (Polysciences, Philadelphia, PA, USA; Cat. no.: WBE20A12E).
- ViiA7 qPCR machine with fast 96-well blocks (ThermoFisher Scientific, Waltham, MA, USA; Cat. no.: 4453535)
- Nanodrop-2000 (ThermoFisher Scientific, Waltham, MA, USA; Cat. no.: ND-2000)
- Vortex mixer (Heidolph, Schwabach, Germany; Cat. no.: 541-10000-00)
- Refrigerated centrifuge with plate adapters (Eppendorf, Hamburg, Germany; Cat. no.: 5811000088 and 5810725003, respectively)
- Sprout® Mini Centrifuge (Heathrow Scientific, Vernon Hills, IL, USA; Cat. no.: HS120301)
- Well calibrated micropipettes (as indicated) (ThermoFisher Scientific, Waltham, MA, USA; Cat. no.: 4700880)
3. Procedure
3.1. DNA Isolation. Time for Completion of 30 Samples: 02:30 h
- Resuspend the blood sample (total 1 mL) thoroughly by inversion and transfer the 900 μL of blood into a fresh 15 mL conical tube.
- Follow the steps as detailed in the QIAamp™ Blood Midi kit handbook. We obtain consistent results with a minimum variation using this kit and recommend the following minor modifications.
- Molecular biology grade ethanol 100%, pure and fresh, is recommended than 96–100% (Qiagen® protocol).
- Use nuclease-free cut filter tips rather than regular filter tips. This allows for easier workflow when dealing with high viscosity blood samples.
- Handling of 30 samples at a time can be a bit tricky. This aspect is described in the higher throughput portion of the kit protocol. We recommend placing all 30 isolation tubes in a single rack and clamping it to another rack with elastic bands. We suggest mixing samples by using a flat head Vortex mixer at the lysis step. The following steps (ethanol addition) could use inversion or vortexing.
- We observe better yield and quality when using a water bath incubator for 70 °C incubation and extending the incubation time to 15 min.
 PAUSE STEP: If using the QIAamp™ kit, elute DNA in buffer AE and aliquot for long-term storage into multiple nuclease-free tubes to be stored at −80 °C.
PAUSE STEP: If using the QIAamp™ kit, elute DNA in buffer AE and aliquot for long-term storage into multiple nuclease-free tubes to be stored at −80 °C. 3.2. DNA Quantitation and Dilution. Time for Completion: 01:00 h for 30 Samples
- Quantify DNA samples and Control DNA on Nanodrop. The 260/280 ratio for isolated DNA using the modified QIAampTM; Blood Midi kit protocol is ≥1.8. DNA quality can be assessed using agarose gel-based separation confirming the absence of any degraded DNA in the form of a smear on the gel (Figure S1).
- Dilute each DNA sample to a required concentration. Here we used three different concentrations during the optimization stage. With the actual experiments (with clinical samples), we use a concentration of 25 ng/μL in a total volume of 8 μL (i.e., 200 ng of total DNA in 8 μL of total volume). Dilutions are performed with the addition of nuclease-free water.
- Spin and store undiluted DNA samples at −80 °C for long-term storage.
PAUSE STEP: Diluted DNA can be stored at −80 °C for up to one week.3.3. Monoplex Real-time PCR. Time for Completion: 04:00 h for 30 samples and Two Genes
3.3.1. Preparation of the Master-Mix
- Create the master-mix as per the volumes of each reagent indicated per reaction (below, Table 1).
![Mps 03 00027 i002]() CRITICAL STEP: It is important to add 5% extra to account for pipetting errors. This ensures that you have enough master-mix for the desired number of reactions that are planned. See an example in Table 1 below.
CRITICAL STEP: It is important to add 5% extra to account for pipetting errors. This ensures that you have enough master-mix for the desired number of reactions that are planned. See an example in Table 1 below. - Reconstitute primers with nuclease-free water to a stock concentration of 5 mM.
- Dilute the telomere A primer stock serially to 100 μM and then to 1μM (working concentration) using nuclease-free water.
- Dilute the telomere B primer stock serially to 100 μM and then to 3μM (working concentration) using nuclease-free water.
- Dilute the hbg1 primer stock serially to 100 μM and then to 3μM (working concentration) using nuclease-free water.
- Dilute the hbg2 primer stock serially to 100 μM and then to 7μM (working concentration) using nuclease-free water.
- Vortex and spin the SYBR green master-mix and primers before addition.
- Prepare two master-mixes, one for telomere PCR that contains telomere A and telomere B primers; whereas another one for single-copy gene PCR contains hbg1 and hbg2 primers.

3.3.2. Plate Setup
- Briefly vortex and centrifuge the master-mix.
- Pipette 9 μL of the above mixture (either for telomere or human β-globin) into each well of a Fast Optical 96-Well Reaction Plate (0.1 mL), then pipette 1 μL of Nuclease-free water (for no template control/NTC), DNA sample or Control DNA (C), in accordance with the 96-well plate map shown below (Figure 1). We recommend having each sample in triplicate, including the NTC and Control DNA.
- There will be two different plates for each sample. One plate containing telomere primers to measure relative telomere abundance and other plates containing human β-globin primers to measure relative single-copy gene abundance.
- Apply an optical adhesive film over the top of the plate, ensuring there is a complete seal and minimizing the generation of any aerosols at this point.
- Centrifuge the plate at 1800 g (3000 rpm) for 5 min at 4 °C. Make sure that there are no air bubbles in these wells. If present, gently flicks to remove bubbles and centrifuge again at 3000 rpm for a further 2 min.
3.3.3. Real-Time PCR
- Insert the plate into the ViiA™ 7 Real-Time PCR System (CONFIRM CORRECT ORIENTATION), load up the Quantstudio Real-time PCR software on the computer and click Experiment Setup.
- Select ViiA7 instrument, Fast 96 Well (0.1 mL) block, Standard Curve, SYBR Green reagents, and FAST options from the setup.
- Enter the target names and sample names and assign correct labels to the wells. Enter reaction volume of 10 μL.
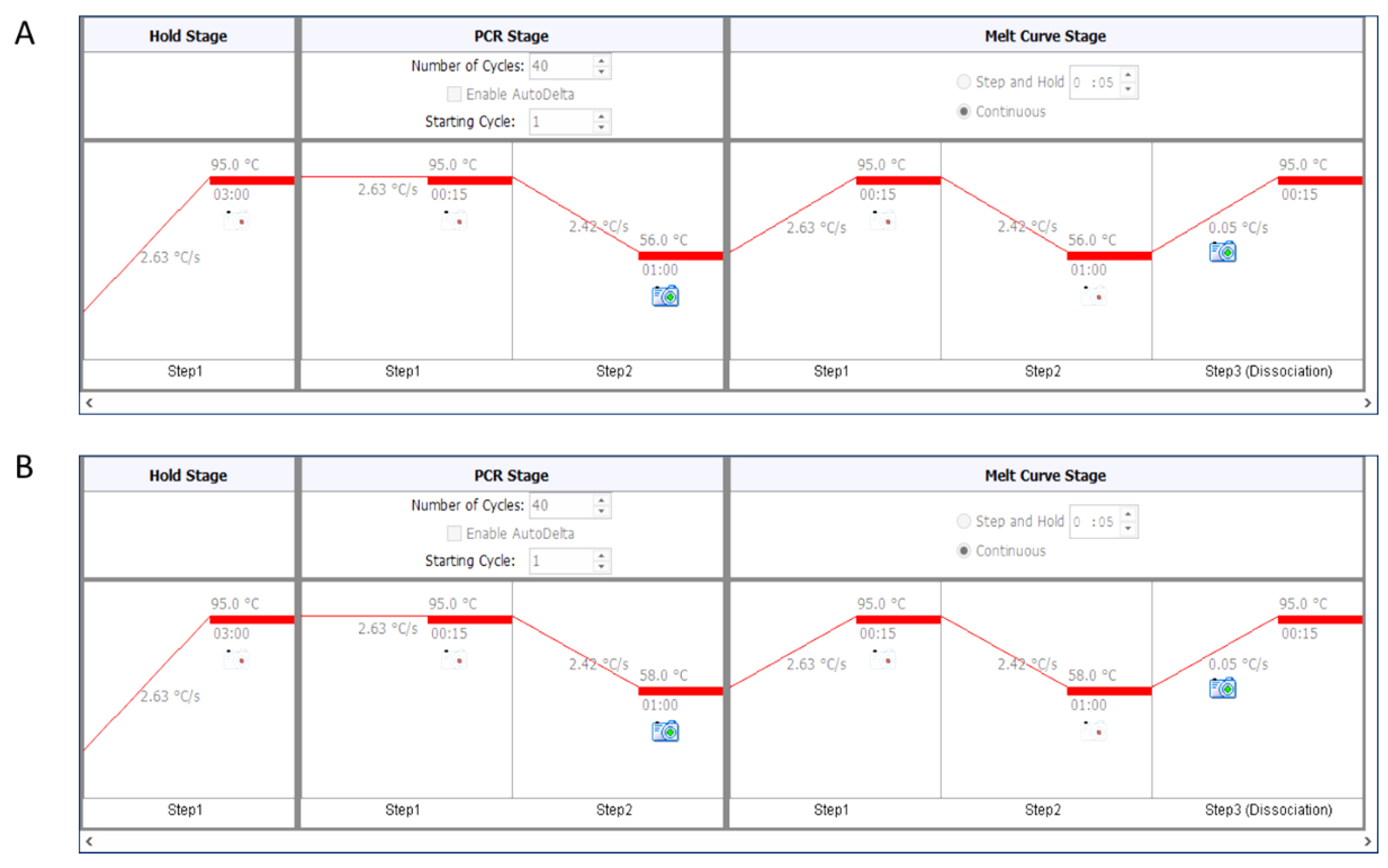
- For telomere PCR, use 56 °C as annealing temperature (Figure 2A).
- For single-copy genes (here human β-globin), use 58 °C as annealing temperature (Figure 2B).
- To start the reaction, save the run and then click START RUN.
3.4. Data Analysis. Time for Completion: 01:00 h
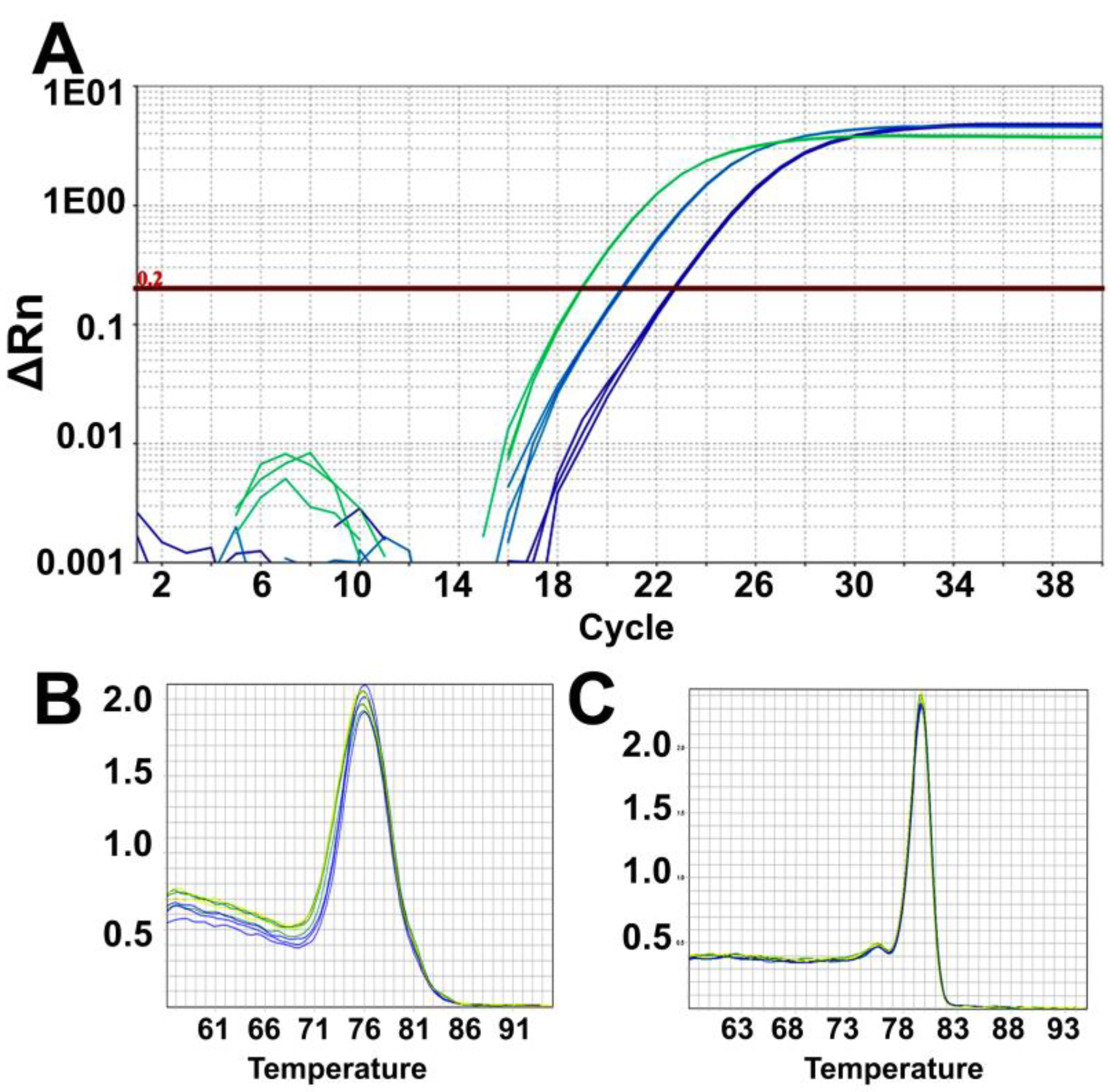
- Analyze the data using the same threshold for all samples in the Quantstudio real-time PCR software and then export the results as an excel sheet. Results are analyzed and plotted using Excel and GraphPad Prism.
![Mps 03 00027 i002]() CRITICAL STEP: It is important to keep the same threshold (such as 0.2) across all the plates from where data is to be analyzed.
CRITICAL STEP: It is important to keep the same threshold (such as 0.2) across all the plates from where data is to be analyzed. - Organize the results as triplicates cycle threshold values (Ct) of telomere and human β-globin genes for each sample, NTC, and Control DNA (Supplementary Table S1).
- Calculate average Ct and standard deviation of each gene for each sample, NTC, and Control DNA.
- Calculate ΔCt for each sample as (Average Ct of hbg)-(Average Ct of telomere). Since telomeres are present in multiple copies in a cell, as against single-copy genes, ΔCt value is positive.
- Similarly, calculate Control DNA ΔCt values.
- Calculate relative telomere length as ∆∆Ct = (Sample Average hbg Ct-Sample Average Telomere Ct)–(Control DNA Average hbg Ct-Control DNA Average Telomere Ct). This value can be represented as the relative telomere length (RTL).
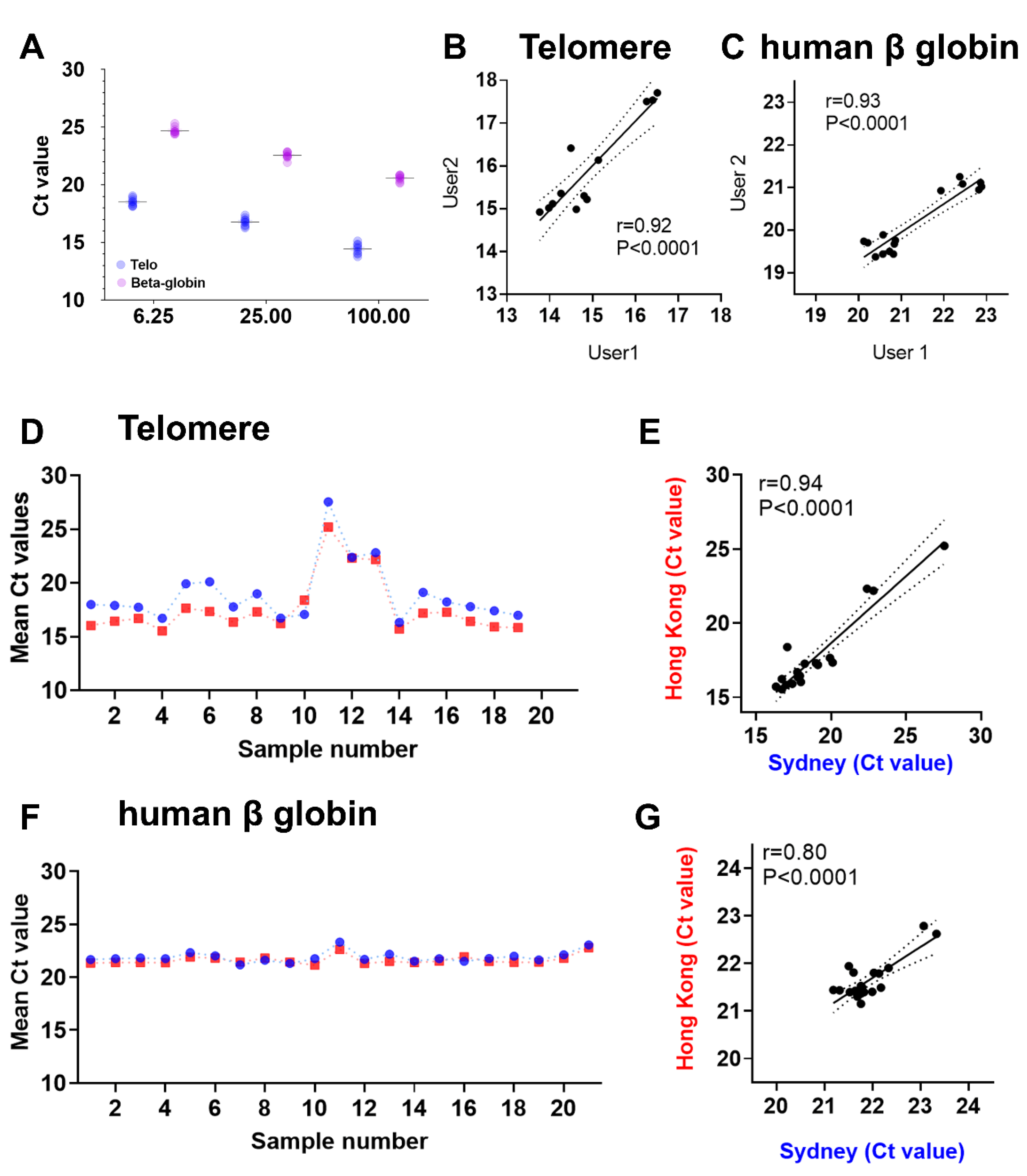
- Calculate the intra-assay CV between the replicate values of Control DNA in the same plate and inter-assay CV between the replicate values of Control DNA from at least three different plates.
4. Results and Discussion
5. Reagents Setup
- All plasticware should be nuclease-free. Never stick your hands into containers of nuclease-free plastic ware. Always pour out the required tubes.
- The use of appropriate PPE is a must.
- Equilibrate blood samples to room temperature (15–25 °C) before starting by placing a batch of 30 samples in a rack at room temperature.
- Follow the DNA isolation protocol provided by the manufacturer. If using the QIAamp™ kit, prepare buffers for DNA isolation as recommended in the QIAamp™ Blood Midi kit handbook and also setup the water bath at 70 °C before initiating DNA isolation.
- All reagents for PCR are stored at −20 °C. Allow samples, primers, and master-mix to thaw thoroughly on ice prior to commencing.
- Master-mix can be aliquoted into smaller volumes (500 μL) to avoid multiple freeze thaws.
- Reconstitute the primers and then dilute them serially to 100 μM first and then to the lower working concentration, as mentioned in the procedures above.
- Make enough volumes of the dilutions for multiple runs in advance. 100 μL of the working concentration primers are required to run one 96-well plate.
- Avoid freeze-thaw of primers and Control DNA samples.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.; Brüderlein, S.; Perner, S.; Waibel, I.; Holdenried, A.; Ciloglu, N.; Hasel, C.; Mattfeldt, T.; Nielsen, K.V.; Möller, P. Sex-specific telomere length profiles and age-dependent erosion dynamics of individual chromosome arms in humans. Cytogenet. Genome Res. 2006, 112, 194–201. [Google Scholar] [CrossRef] [PubMed]
- McCord, R.P.; Nazario-Toole, A.; Zhang, H.; Chines, P.S.; Zhan, Y.; Erdos, M.R.; Collins, F.S.; Dekker, J.; Cao, K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013, 23, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Savage, S.A.; Giri, N.; Baerlocher, G.M.; Orr, N.; Lansdorp, P.M.; Alter, B.P. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 2008, 82, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, M.; Pusceddu, I.; März, W.; Herrmann, W. Telomere biology and age-related diseases. Clin. Chem. Lab. Med. 2018, 56, 1210–1222. [Google Scholar] [CrossRef]
- Shammas, M.A. Telomeres, lifestyle, cancer, and aging. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Montpetit, A.J.; Alhareeri, A.A.; Montpetit, M.; Starkweather, A.R.; Elmore, L.W.; Filler, K.; Mohanraj, L.; Burton, C.W.; Menzies, V.S.; Lyon, D.E.; et al. Telomere length: A review of methods for measurement. Nurs. Res. 2014, 63, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, T.P.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cawthon, R.M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002, 30, e47. [Google Scholar] [CrossRef] [PubMed]
- Vasilishina, A.; Kropotov, A.; Spivak, I.; Bernadotte, A. Relative Human Telomere Length Quantification by Real-Time PCR. Methods Mol. Biol. 2019, 1896, 39–44. [Google Scholar] [PubMed]
- Lin, J.; Smith, D.L.; Esteves, K.; Drury, S. Telomere length measurement by qPCR-Summary of critical factors and recommendations for assay design. Psychoneuroendocrinology 2019, 99, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.M.; Johnson, R.A.; Litzelman, K.; Skinner, H.G.; Seo, S.; Engelman, C.D.; Vanderboom, R.J.; Kimmel, G.W.; Gangnon, R.E.; Riegert-Johnson, D.L.; et al. Telomere length varies by DNA extraction method: Implications for epidemiologic research. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2047–2054. [Google Scholar] [CrossRef] [Green Version]
- Denham, J.; Marques, F.Z.; Charchar, F.J. Leukocyte telomere length variation due to DNA extraction method. BMC Res. Notes 2014, 7, 877. [Google Scholar] [CrossRef] [Green Version]
- Raschenberger, J.; Lamina, C.; Haun, M.; Kollerits, B.; Coassin, S.; Boes, E.; Kedenko, L.; Köttgen, A.; Kronenberg, F. Influence of DNA extraction methods on relative telomere length measurements and its impact on epidemiological studies. Sci. Rep. 2016, 6, 25398. [Google Scholar] [CrossRef] [Green Version]
- Dagnall, C.L.; Hicks, B.; Teshome, K.; Hutchinson, A.A.; Gadalla, S.M.; Khincha, P.P.; Yeager, M.; Savage, S.A. Effect of pre-analytic variables on the reproducibility of qPCR relative telomere length measurement. PLoS ONE 2017, 12, e0184098. [Google Scholar] [CrossRef]
- Zanet, D.L.; Saberi, S.; Oliveira, L.; Sattha, B.; Gadawski, I.; Côté, H.C. Blood and dried blood spot telomere length measurement by qPCR: Assay considerations. PLoS ONE 2013, 8, e57787. [Google Scholar] [CrossRef]
- Jimenez, K.M.; Forero, D.A. Effect of master mixes on the measurement of telomere length by qPCR. Mol. Biol. Rep. 2018, 45, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ruiz, C.M.; Baird, D.; Roger, L.; Boukamp, P.; Krunic, D.; Cawthon, R.; Dokter, M.M.; van der Harst, P.; Bekaert, S.; de Meyer, T.; et al. Reproducibility of telomere length assessment: An international collaborative study. Int. J. Epidemiol. 2015, 44, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, K.; Passos, J.F.; Olijslagers, S.; Saretzki, G.; Martin-Ruiz, C.; von Zglinicki, T. Premature senescence of mesothelial cells is associated with non-telomeric DNA damage. Biochem. Biophys. Res. Commun. 2007, 362, 707–711. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.; Wong, J.Y.; Michaud, D.; Hunter, D.J.; De Vivo, I. Telomere length, cigarette smoking, and bladder cancer risk in men and women. Cancer Epidemiol. Biomark. Prev. 2007, 16, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Slykerman, R.F.; Joglekar, M.V.; Hardikar, A.A.; Satoor, S.N.; Thompson, J.M.; Jenkins, A.; Mitchell, E.A.; Murphy, R. Maternal stress during pregnancy and small for gestational age birthweight are not associated with telomere length at 11years of age. Gene 2019, 694, 97–101. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Volume/Reaction (μL) | Volume for 96 Reactions (μL) |
|---|---|---|
| 2X Fast SYBR® Green Master Mix | 5 | 504 |
| Telomere A OR hbg1 primer (working concentration) | 1 | 100.8 |
| Telomere B OR hbg2 primer (working concentration) | 1 | 100.8 |
| Nuclease-free water | 2 | 201.6 |
| TOTAL | 9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joglekar, M.V.; Satoor, S.N.; Wong, W.K.M.; Cheng, F.; Ma, R.C.W.; Hardikar, A.A. An Optimised Step-by-Step Protocol for Measuring Relative Telomere Length. Methods Protoc. 2020, 3, 27. https://doi.org/10.3390/mps3020027
Joglekar MV, Satoor SN, Wong WKM, Cheng F, Ma RCW, Hardikar AA. An Optimised Step-by-Step Protocol for Measuring Relative Telomere Length. Methods and Protocols. 2020; 3(2):27. https://doi.org/10.3390/mps3020027
Chicago/Turabian StyleJoglekar, Mugdha V., Sarang N. Satoor, Wilson K.M. Wong, Feifei Cheng, Ronald C.W. Ma, and Anandwardhan A. Hardikar. 2020. "An Optimised Step-by-Step Protocol for Measuring Relative Telomere Length" Methods and Protocols 3, no. 2: 27. https://doi.org/10.3390/mps3020027