
Anticancer Activity of Thiophene Carboxamide Derivatives as CA-4 Biomimetics: Synthesis, Biological Potency, 3D Spheroid Model, and Molecular Dynamics Simulation
 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
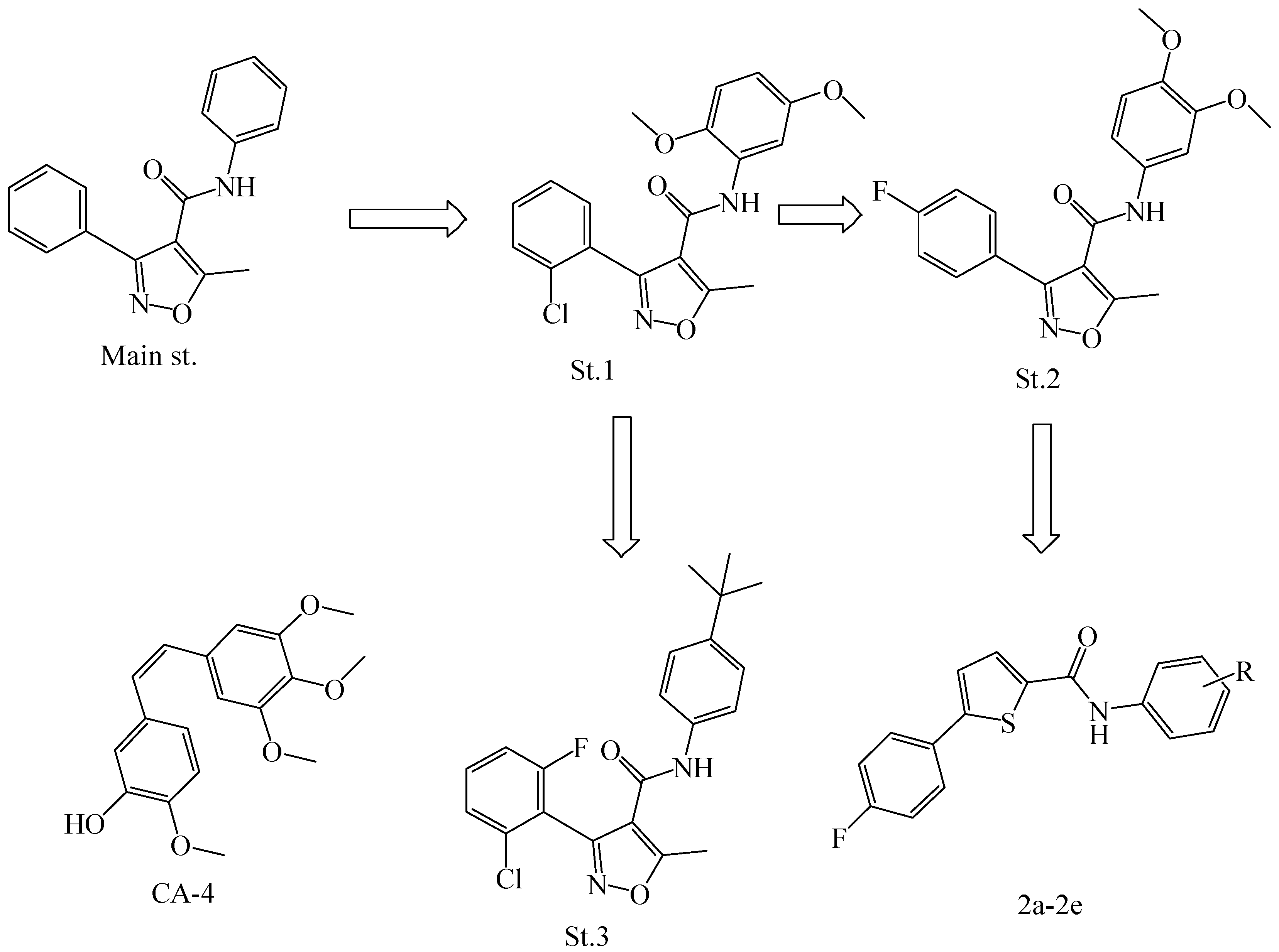
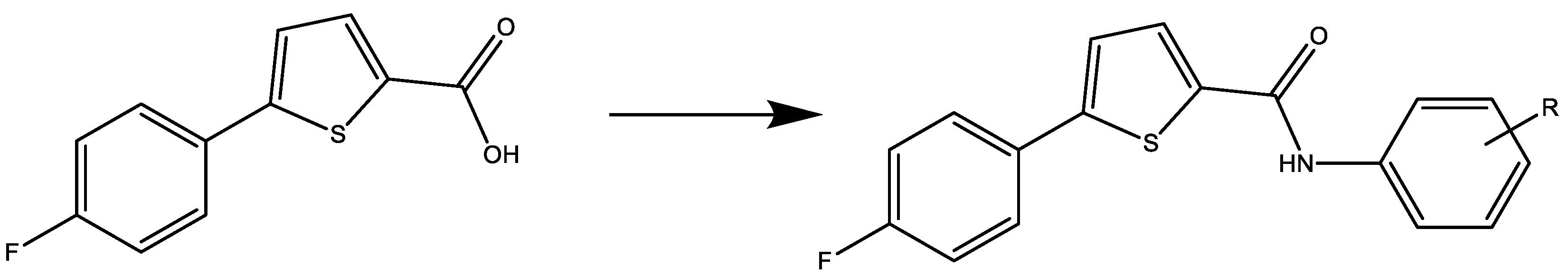
2.1. Chemistry
General Procedure for the Synthesis of thiophene-Carboxamide (2a–2e)
2.2. Biological Methods
2.2.1. Cytotoxicity Method
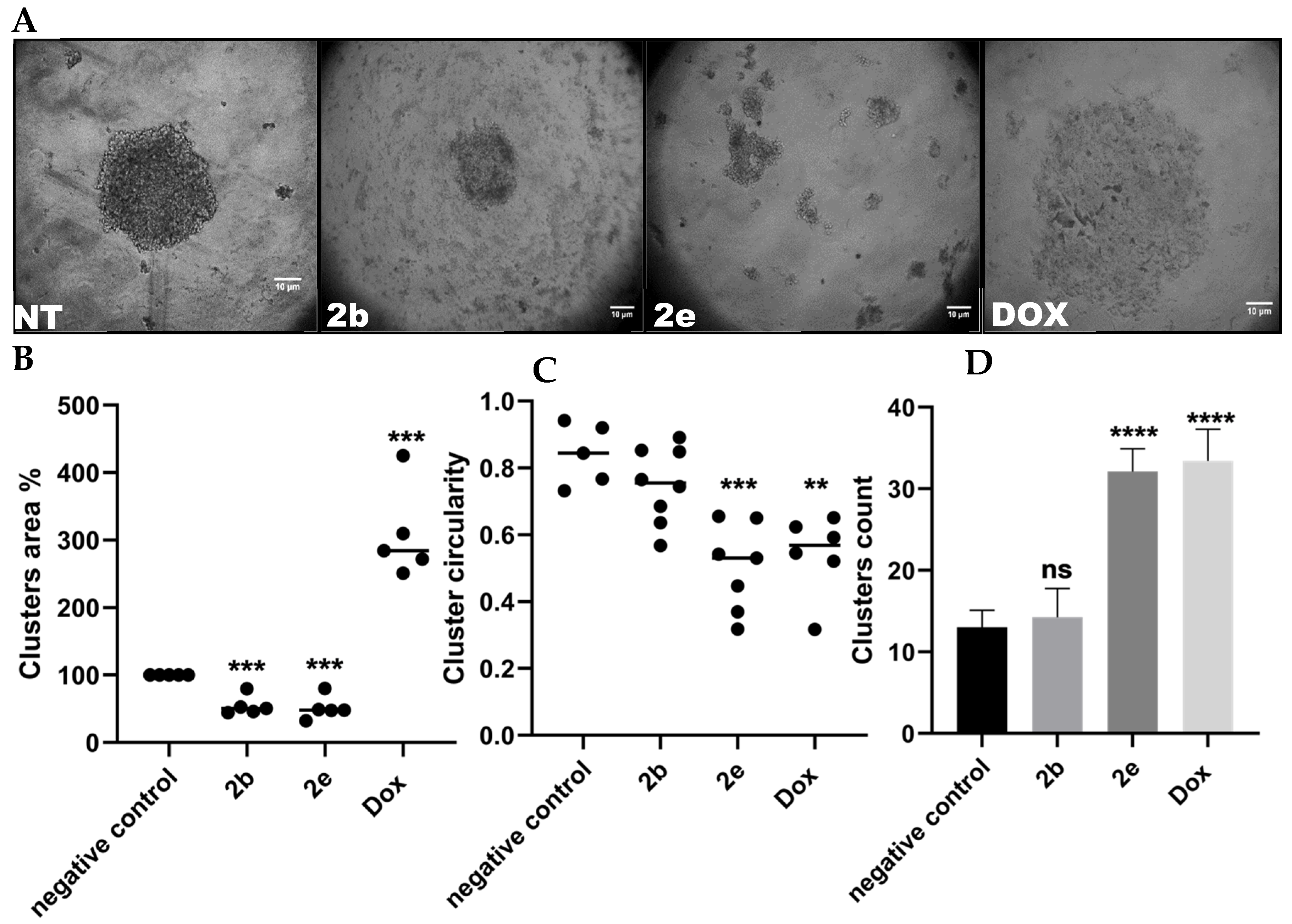
2.2.2. Hepatocellular Carcinoma Spheroids Formation Test
2.3. Bioinformatic Investigations
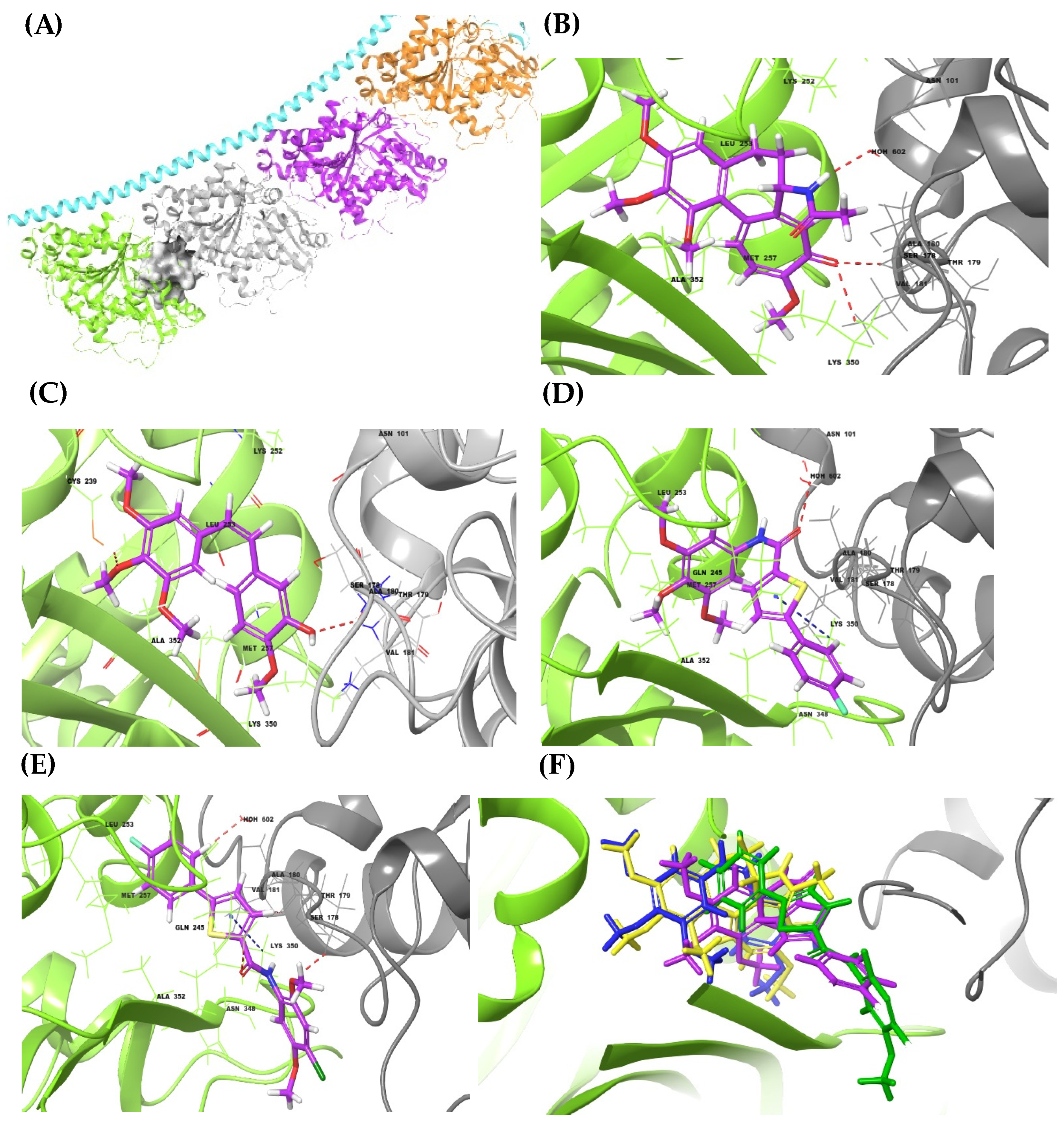
2.3.1. Molecular Docking Studies
2.3.2. Free Energy Calculations Using Prime MM-GBSA
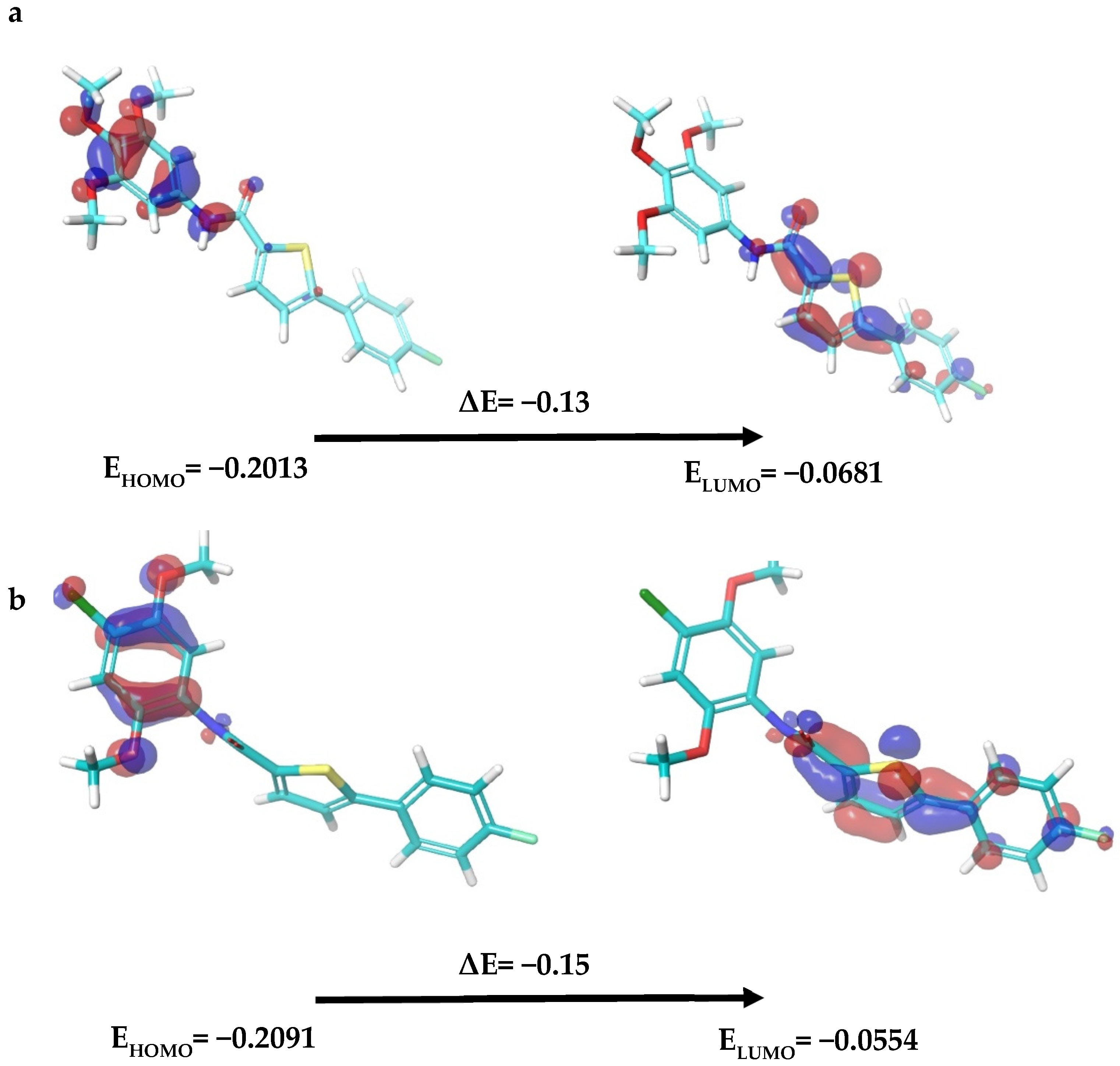
2.3.3. Density Functional Theory (DFT) Analysis
2.3.4. Molecular Dynamics (MD) Simulations
2.3.5. Ligand-Based ADME/Toxicity Prediction
2.4. Statistical Analysis
3. Results
3.1. Chemistry
3.2. Chemical Characterization
- 5-(4-fluorophenyl)-N-phenylthiophene-2-carboxamide (2a)
- 5-(4-fluorophenyl)-N-(3,4,5-trimethoxyphenyl) thiophene-2-carboxamide (2b)
- N-(4-(tert-butyl) phenyl)-5-(4-fluorophenyl) thiophene-2-carboxamide (2c)
- N-(3,5-dimethoxyphenyl)-5-(4-fluorophenyl) thiophene-2-carboxamide (2d)
- N-(4-chloro-2,5-dimethoxyphenyl)-5-(4-flurophenyl)thiophene-2-carboxamide (2e)
3.3. Biological Evaluations
3.3.1. Cytotoxic Evaluation of the Compounds 2a–2e MTS Assay Results
3.3.2. Dimensional (3D) Hepatocellular Spheroids’ Formation
3.4. Bioinformatics Studies
3.4.1. Molecular Docking Simulations and MM-GBSA
3.4.2. Density Functional Theory Analysis
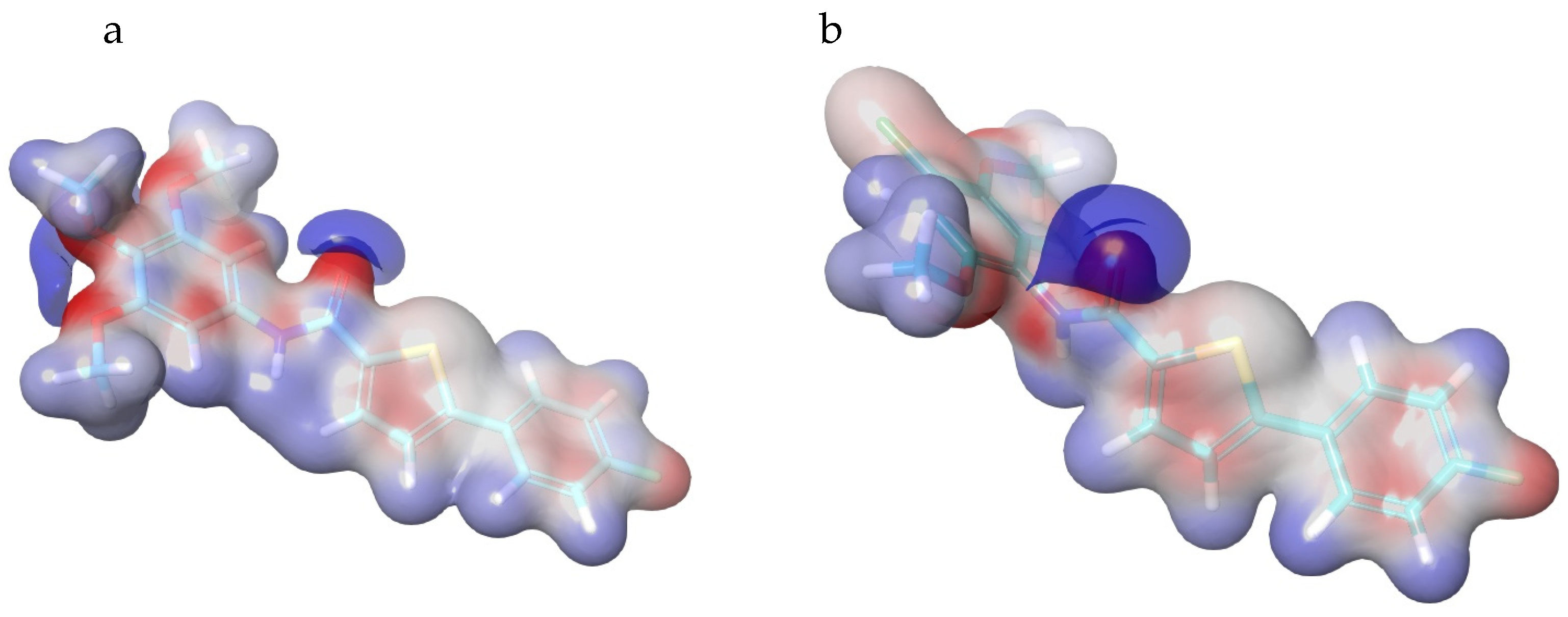
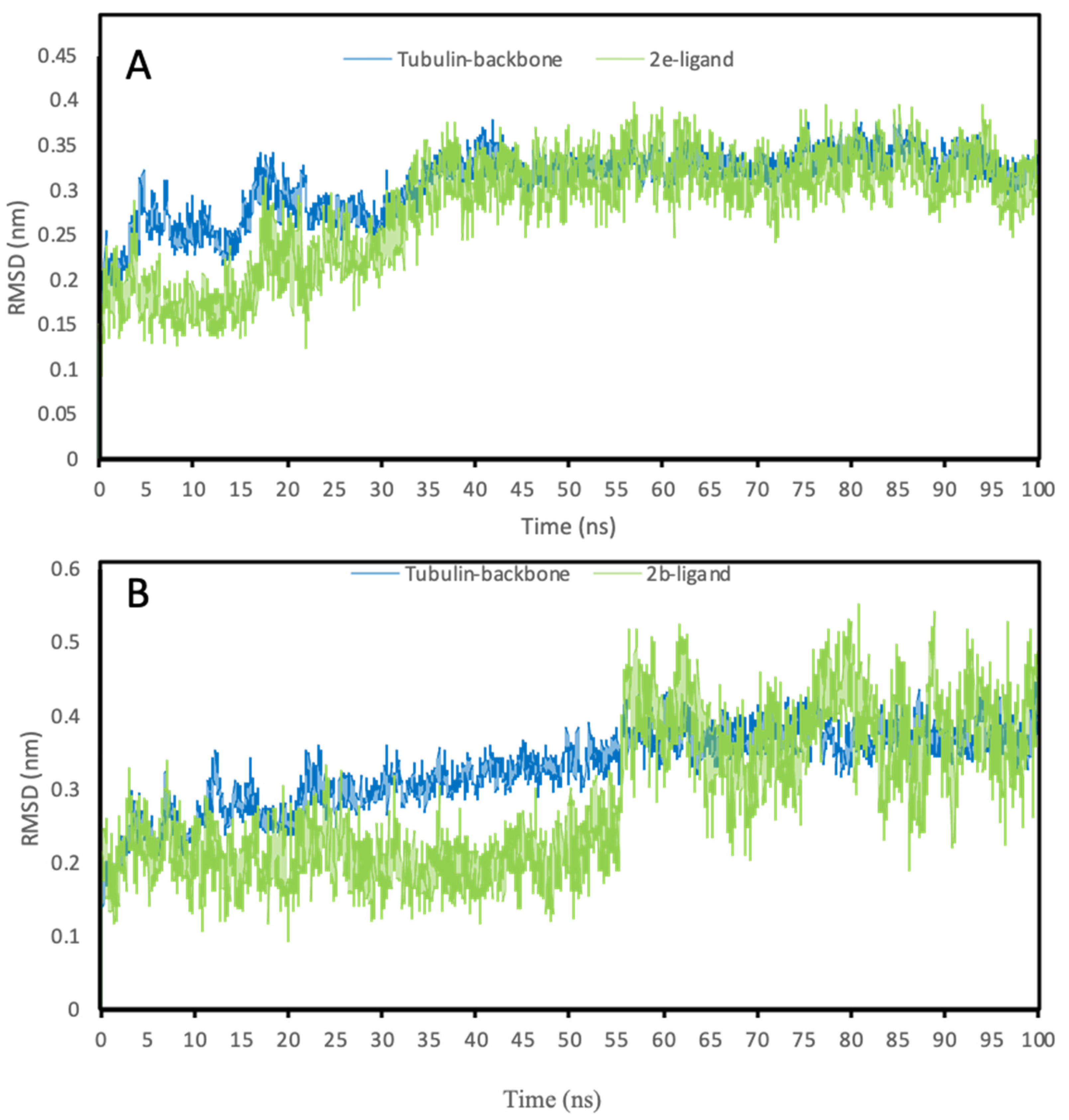
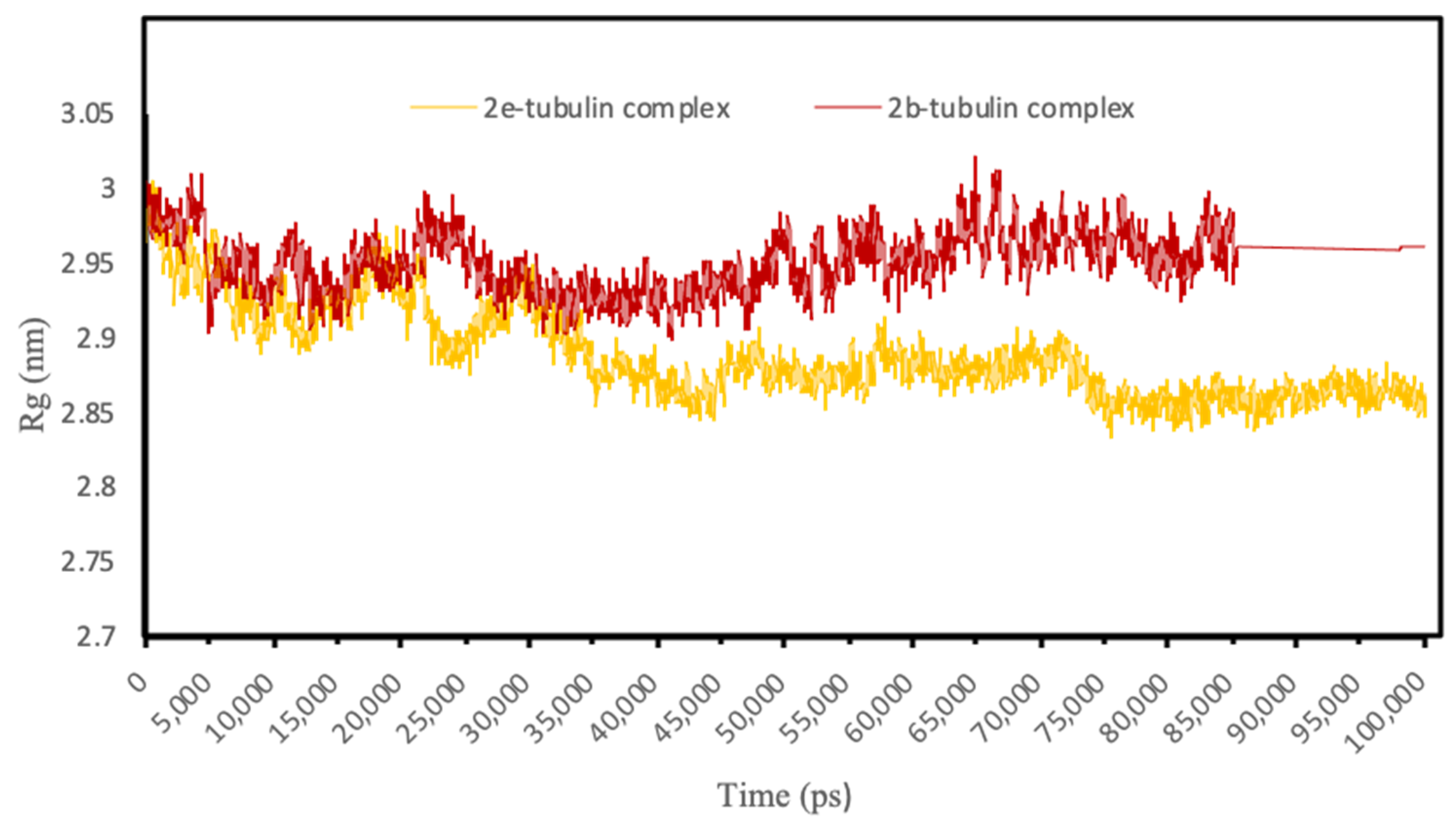
3.4.3. Molecular Dynamics Simulations
3.4.4. ADME-T Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global cancer observatory: Cancer today. Int. Agency Res. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Hawash, M. Highlights on Specific Biological Targets; Cyclin-Dependent Kinases, Epidermal Growth Factor Receptors, Ras Protein, and Cancer Stem Cells in Anticancer Drug Development. Drug Res. 2019, 69, 471–478. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Ju, D.T.; Chang, C.F.; Muralidhar Reddy, P.; Velmurugan, B.K. A review on the effects of current chemotherapy drugs and natural agents in treating non-small cell lung cancer. Biomedicine 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, A.M.; Hawash, M.; Amer, J.; Jarrar, A.; Qadri, S.; Alnimer, I.; Sharaf, A.; Zalmoot, R.; Hammoudie, O.; Hameedi, S.; et al. Synthesis and Biological Evaluation of Novel Isoxazole-Amide Analogues as Anticancer and Antioxidant Agents. Biomed Res. Int. 2021, 2021, 6633297. [Google Scholar] [CrossRef]
- Baytas, S.N.; Inceler, N.; Yılmaz, A.; Olgac, A.; Menevse, S.; Banoglu, E.; Hamel, E.; Bortolozzi, R.; Viola, G. Synthesis, biological evaluation and molecular docking studies of trans-indole-3-acrylamide derivatives, a new class of tubulin polymerization inhibitors. Bioorg. Med. Chem. 2014, 22, 3096–3104. [Google Scholar] [CrossRef] [Green Version]
- Jaradat, N.A.; Al-lahham, S.; Zaid, A.N.; Hussein, F.; Issa, L.; Abualhasan, M.N.; Hawash, M.; Yahya, A.; Shehadi, O.; Omair, R. Carlina curetum plant phytoconstituents, enzymes inhibitory and cytotoxic activity on cervical epithelial carcinoma and colon cancer cell lines. Eur. J. Integr. Med. 2019, 30, 100933. [Google Scholar] [CrossRef]
- Baytas, S.N.; Inceler, N.; Yılmaz, A. Synthesis, cytotoxicity, and molecular properties prediction of novel 1,3-diarylpyrazole derivatives. Med. Chem. Res. 2013, 22, 4893–4908. [Google Scholar] [CrossRef]
- Hawash, M. Recent Advances of Tubulin Inhibitors Targeting the Colchicine Binding Site for Cancer Therapy. Biomolecules 2022, 12, 1843. [Google Scholar] [CrossRef]
- BAYTAŞ, S.; Dural, N.N.T.; ÖZKAN, Y.; ŞİMŞEK, H.B.; Gürsel, T.; Ünlü, S. Synthesis, anti-inflammatory, antiplatelet and in silico evaluations of (E)-3-(3-(2, 3-dihydro-3-methyl-2-oxo-3H-benzoxazole-6-yl)-1-phenyl-1H-pyrazole-4-yl) acrylamides. Turk. J. Chem. 2012, 36, 367–382. [Google Scholar] [CrossRef]
- Hawash, M.; Jaradat, N.; Abualhasan, M.; Qaoud, M.T.; Joudeh, Y.; Jaber, Z.; Sawalmeh, M.; Zarour, A.; Mousa, A.; Arar, M. Molecular docking studies and biological evaluation of isoxazole-carboxamide derivatives as COX inhibitors and antimicrobial agents. 3 Biotech 2022, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Inceler, N.; Yılmaz, A.; Baytas, S.N. Synthesis of ester and amide derivatives of 1-phenyl-3-(thiophen-3-yl)-1 H-pyrazole-4-carboxylic acid and study of their anticancer activity. Med. Chem. Res. 2013, 22, 3109–3118. [Google Scholar] [CrossRef]
- Inceler, N.; Ozkan, Y.; Turan, N.N.; Kahraman, D.C.; Cetin-Atalay, R.; Baytas, S.N. Design, synthesis and biological evaluation of novel 1, 3-diarylpyrazoles as cyclooxygenase inhibitors, antiplatelet and anticancer agents. MedChemComm 2018, 9, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hashem, A.A. Synthesis of new pyrazoles, oxadiazoles, triazoles, pyrrolotriazines, and pyrrolotriazepines as potential cytotoxic agents. J. Heterocycl. Chem. 2021, 58, 805–821. [Google Scholar] [CrossRef]
- Gomha, S.M.; Salah, T.A.; Abdelhamid, A.O. Synthesis, characterization, and pharmacological evaluation of some novel thiadiazoles and thiazoles incorporating pyrazole moiety as anticancer agents. Mon. Für Chem.-Chem. Mon. 2015, 146, 149–158. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals: Miniperspective. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Carbone, D.; Vestuto, V.; Ferraro, M.R.; Ciaglia, T.; Pecoraro, C.; Sommella, E.; Cascioferro, S.; Salviati, E.; Novi, S.; Tecce, M.F. Metabolomics-assisted discovery of a new anticancer GLS-1 inhibitor chemotype from a nortopsentin-inspired library: From phenotype screening to target identification. Eur. J. Med. Chem. 2022, 234, 114233. [Google Scholar] [CrossRef]
- Pecoraro, C.; Parrino, B.; Cascioferro, S.; Puerta, A.; Avan, A.; Peters, G.J.; Diana, P.; Giovannetti, E.; Carbone, D. A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules 2021, 27, 19. [Google Scholar] [CrossRef]
- Hawash, M.; Kahraman, D.C.; Olgac, A.; Ergun, S.G.; Hamel, E.; Cetin-Atalay, R.; Baytas, S.N. Design and Synthesis of Novel Substituted Indole-acrylamide Derivatives and Evaluation of Their Anti-Cancer Activity as Potential Tubulin-Targeting Agents. J. Mol. Struct. 2022, 1254, 132345. [Google Scholar] [CrossRef]
- Mishra, R.; Kumar, N.; Mishra, I.; Sachan, N. A review on anticancer activities of thiophene and its analogs. Mini Rev. Med. Chem. 2020, 20, 1944–1965. [Google Scholar] [CrossRef]
- Pathania, S.; Chawla, P.A. Thiophene-based derivatives as anticancer agents: An overview on decade’s work. Bioorg. Chem. 2020, 101, 104026. [Google Scholar]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.D.; Rathod, P.D.; Xavier, F.P.; Padh, H.; Sudarsanam, V.; Vasu, K.K. Tetra substituted thiophenes as anti-inflammatory agents: Exploitation of analogue-based drug design. Bioorg. Med. Chem. 2005, 13, 6685–6692. [Google Scholar] [CrossRef] [PubMed]
- Tehranchian, S.; Akbarzadeh, T.; Fazeli, M.R.; Jamalifar, H.; Shafiee, A. Synthesis and antibacterial activity of 1-[1, 2, 4-triazol-3-yl] and 1-[1, 3, 4-thiadiazol-2-yl]-3-methylthio-6, 7-dihydrobenzo [c] thiophen-4 (5H) ones. Bioorg. Med. Chem. Lett. 2005, 15, 1023–1025. [Google Scholar] [CrossRef]
- Russell, R.K.; Press, J.B.; Rampulla, R.A.; McNally, J.J.; Falotico, R.; Keiser, J.A.; Bright, D.A.; Tobia, A. Thiophene systems. 9. Thienopyrimidinedione derivatives as potential antihypertensive agents. J. Med. Chem. 1988, 31, 1786–1793. [Google Scholar] [CrossRef]
- Chen, Z.; Ku, T.C.; Seley-Radtke, K.L. Thiophene-expanded guanosine analogues of gemcitabine. Bioorg. Med. Chem. Lett. 2015, 25, 4274–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, C.D.; Horsman, M.R.; Paul, E.G.; Kristjansen, P.E.G. Early Effects of Combretastatin-A4 Disodium Phosphate on Tumor Perfusion and Interstitial Fluid Pressure. Neoplasia 2007, 9, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, S.T.; Oltra, S.D.; Falomir, E.; Murga, J.; Carda, M.; Marco, J.A. Synthesis of combretastatin A-4 O-alkyl derivatives and evaluation of their cytotoxic, antiangiogenic and antitelomerase activity. Bioorg. Med. Chem. J. 2013, 21, 7267–7274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Zhang, D.; Jin, Q.; Jiang, C.; Wang, C.; Li, J.; Peng, F.; Huang, D.; Zhang, J.; Song, S. Combretastatin-A4 phosphate improves the distribution and antitumor efficacy of albumin-bound paclitaxel in W256 breast carcinoma model. Oncotarget Impact J. 2016, 7, 58133–58141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, P.; Zweifel, M.; Padhani, A.R.; Koh, D.M.; Ng, M.; Collins, D.J.; Harris, A.; Carden, C.; Smythe, J.; Fisher, N.; et al. Phase I Trial of Combretastatin A4 Phosphate (CA4P) in Combination with Bevacizumab in Patients with Advanced Cancer. Clin. Cancer Res. 2012, 18, 3428–3439. [Google Scholar] [CrossRef] [Green Version]
- Simoni, D.; Romagnoli, R.; Baruchello, R.; Rondanin, R.; Rizzi, M.; Pavani, M.G.; Alloatti, D.; Giannini, G.; Marcellini, M.; Riccioni, T. Novel combretastatin analogues endowed with antitumor activity. J. Med. Chem. 2006, 49, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.P.; Rosen, M.; Sun, W.; Gallagher, M.; Haller, D.G.; Vaughn, D.; Giantonio, B.; Zimmer, R.; Petros, W.P.; Stratford, M. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: Magnetic resonance imaging evidence for altered tumor blood flow. J. Clin. Oncol. 2003, 21, 4428–4438. [Google Scholar] [CrossRef] [PubMed]
- Hawash, M.; Jaradat, N.; Abualhasan, M.; Qneibi, M.; Rifai, H.; Saqfelhait, T.; Shqirat, Y.; Nazal, A.; Omarya, S.; Ibrahim, T.; et al. Evaluation of cytotoxic, COX inhibitory, and antimicrobial activities of novel isoxazole-carboxamide derivatives. Lett. Drug Des. Discov. 2022, 19, 1. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef]
- Aprile, S.; Del Grosso, E.; Tron, G.C.; Grosa, G. In vitro metabolism study of combretastatin A-4 in rat and human liver microsomes. Drug Metab. Dispos. 2007, 35, 2252–2261. [Google Scholar] [CrossRef] [Green Version]
- Hawash, M.; Jaradat, N.; Abualhasan, M.; Amer, J.; Levent, S.; Issa, S.; Ibrahim, S.; Ayaseh, A.; Shtayeh, T.; Mousa, A. Synthesis, chemo-informatics, and anticancer evaluation of fluorophenyl-isoxazole derivatives. Open Chem. 2021, 19, 855–863. [Google Scholar] [CrossRef]
- Hawash, M.; Jaradat, N.; Bawwab, N.; Salem, K.; Arafat, H.; Hajyousef, Y.; Shtayeh, T.; Sobuh, S. Design, synthesis, and biological evaluation of phenyl-isoxazole-carboxamide derivatives as anticancer agents. Heterocycl. Commun. 2021, 27, 133–141. [Google Scholar] [CrossRef]
- Shi, M.; Ho, K.; Keating, A.; Shoichet, M.S. Doxorubicin-conjugated immuno-nanoparticles for intracellular anticancer drug delivery. Adv. Funct. Mater. 2009, 19, 1689–1696. [Google Scholar] [CrossRef]
- Amer, J.; Salhab, A.; Jaradat, N.; Abdallah, S.; Aburas, H.; Hattab, S.; Ghanim, M.; Alqub, M. Gundelia tournefortii inhibits hepatocellular carcinoma progression by lowering gene expression of the cell cycle and hepatocyte proliferation in immunodeficient mice. Biomed. Pharm. 2022, 156, 113885. [Google Scholar] [CrossRef]
- Schrödinger, L. The PyMol Molecular Graphics System, Version, 2017. 2; Schrödinger, LLC.: New York, NY, USA, 2017. [Google Scholar]
- Release, S. 4: Schrödinger Suite 2017-4 Protein Preparation Wizard; Epik, Schrödinger, LLC.: New York, NY, USA; Impact Schrödinger LLC.: New York, NY, USA, 2017. [Google Scholar]
- Release, S. 3: LigPrep; Schrödinger, LLC.: New York, NY, USA, 2021. [Google Scholar]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Jaguar, V. 9.1 Schrödinger Suite Release 2016-1; Schrödinger LLC.: New York, NY, USA, 2016. [Google Scholar]
- Gill, P.M.; Johnson, B.G.; Pople, J.A.; Frisch, M.J. The performance of the Becke—Lee—Yang—Parr (B—LYP) density functional theory with various basis sets. Chem. Phys. Lett. 1992, 197, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- de Groot, R.A.; Nadrchal, J. Physics Computing’92: Proceedings of the 4th International Conference. In Proceedings of the Physics Computing’92: Proceedings of the 4th International Conference, Prague, Czechoslovakia, 24–28 August 1992; Nadrchal, J., Ed.; World Scientific Publishing Co., Pte. Ltd.: Singapore. [Google Scholar]
- Van Aalten, D.M.; Bywater, R.; Findlay, J.B.; Hendlich, M.; Hooft, R.W.; Vriend, G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput.-Aided Mol. Des. 1996, 10, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the reliability of QikProp. Correlation between experimental and predicted values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Hawash, M.; Eid, A.M.; Jaradat, N.; Abualhasan, M.; Amer, J.; Naser Zaid, A.; Draghmeh, S.; Daraghmeh, D.; Daraghmeh, H.; Shtayeh, T.; et al. Synthesis and Biological Evaluation of Benzodioxole Derivatives as Potential Anticancer and Antioxidant agents. Heterocycl. Commun. 2020, 26, 157–167. [Google Scholar] [CrossRef]
- Hawash, M.; Kahraman, D.C.; Cetin-Atalay, R.; Baytas, S. Induction of Apoptosis in Hepatocellular Carcinoma Cell Lines by Novel Indolylacrylamide Derivatives: Synthesis and Biological Evaluation. Chem. Biodivers. 2021, 18, e2001037. [Google Scholar] [CrossRef]
- Masuo, K.; Chen, R.; Yogo, A.; Sugiyama, A.; Fukuda, A.; Masui, T.; Uemoto, S.; Seno, H.; Takaishi, S. SNAIL2 contributes to tumorigenicity and chemotherapy resistance in pancreatic cancer by regulating IGFBP2. Cancer Sci. 2021, 112, 4987. [Google Scholar] [CrossRef]
- Melnik, D.; Sahana, J.; Corydon, T.J.; Kopp, S.; Nassef, M.Z.; Wehland, M.; Infanger, M.; Grimm, D.; Krüger, M. Dexamethasone inhibits spheroid formation of thyroid cancer cells exposed to simulated microgravity. Cells 2020, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Deezagi, A.; Safari, N. Rosuvastatin inhibit spheroid formation and epithelial–mesenchymal transition (EMT) in prostate cancer PC-3 cell line. Mol. Biol. Rep. 2020, 47, 8727–8737. [Google Scholar] [CrossRef] [PubMed]
- Boylan, K.L.; Manion, R.D.; Shah, H.; Skubitz, K.M.; Skubitz, A.P. Inhibition of ovarian cancer cell spheroid formation by synthetic peptides derived from Nectin-4. Int. J. Mol. Sci. 2020, 21, 4637. [Google Scholar] [CrossRef] [PubMed]
- Olatoke, S.; Agodirin, O.; Habeeb, O.; Akande, H. Relationship between tumour size and response to neoadjuvant chemotherapy among breast cancer patients in a tertiary center in Nigeria. Malawi Med. J. 2018, 30, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Crisan, L.; Borota, A.; Bora, A.; Pacureanu, L. Diarylthiazole and diarylimidazole selective COX-1 inhibitor analysis through pharmacophore modeling, virtual screening, and DFT-based approaches. Struct. Chem. 2019, 30, 2311–2326. [Google Scholar] [CrossRef]
- Fernández, I.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory: Understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef]
- Asiri, A.M.; Karabacak, M.; Kurt, M.; Alamry, K.A. Synthesis, molecular conformation, vibrational and electronic transition, isometric chemical shift, polarizability and hyperpolarizability analysis of 3-(4-Methoxy-phenyl)-2-(4-nitro-phenyl)-acrylonitrile: A combined experimental and theoretical analysis. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 82, 444–455. [Google Scholar] [CrossRef]
- Kenny, P.W. Hydrogen bonding, electrostatic potential, and molecular design. J. Chem. Inf. Model. 2009, 49, 1234–1244. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| IC50 (µM) | ||||||
| Cell line | 2a | 2b | 2c | 2d | 2e | DOX |
| R | H | 3,4,5-tri-MeO | 4-t-butyl | 3,5-di-MeO | 2,5-diMeO-4-CL | - |
| Hep3B | 28.02 ± 2.05 | 5.46 ± 1.47 | 189.43 ± 2.78 | 8.85 ± 1.45 | 12.58 ± 2.57 | 1.21 ± 1.0 |
| B16F1 | Ni | Ni | Ni | Ni | 112.11 ± 1.99 | <1 |
| Colo205 | Ni | 35.08 ± 2.05 | Ni | Ni | 74.82 ± 1.45 | <1 |
| HepG2 | 283.67 ± 1.79 | >300 | >300 | >300 | 18.19 ± 0.78 | 1.598 ± 0.67 |
| CaCo-2 | Ni | Ni | Ni | Ni | 82.89 ± 2.45 | <1 |
| HeLa | >300 | 205.33 | >300 | >300 | 39.58 ± 1.88 | 1.55 ± 1.35 |
| MCF-7 | >300 | 29.74 ± 2.74 | Ni | >300 | 42.50 ± 2.59 | <1 |
| LX-2 | Ni | Ni | Ni | Ni | Ni | <1 |
| Hek293t | Ni | 278.13 ± 2.75 | Ni | Ni | 105.23 ± 1.08 | 1.24 ± 0.87 |
| Name | H. Bs | π-Cation | HPHO | Docking Score | MM-GBSA (ΔG) |
|---|---|---|---|---|---|
| 2b | C:ASN 101 | D:LYS 350 | D:LEU 248, D: LEU 255, D: ALA 316 | −6.13 | −36.02 |
| 2e | D:GLN 245, C:SER 178 | D:LYS 350 | D:LEU 246, D: ALA 148, D:LYS 252, D:LYS 350 | −6.46 | −41.47 |
| Colchicine | C:VAL 181, C:ASN 101, D:LYS 350 | D:ALA 248, D:LYS 252, D:LEU 253, D:ASN 256, D:LYS 350, | −9.23 | −42.75 | |
| CA-4 | D:CYS 239, C:VAL 181 | D:LEU 246, D:LEU 253, D:ASN 256 | −8.29 | −46.72 |
| Mol._Mw | PSA | Percent Human Oral Absorption | Dipole | QPlogPo/w | QPlogPoct++ | QPlogBB | QPlogS | QPlogKhsa | SASA | FOSA | FISA | PISA | WPSA | Volume | QPpolrz | QPPCaco | #metab⁑ | QPlogHERG | Rule of three | Rule of five | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | 297.3 | 35.6 | 100 | 5.66 | 4.5 | 14.7 | 0.17 | −5.2 | 0.60 | 558.7 | 0.0 | 47.5 | 433.5 | 77.6 | 939.0 | 34.28 | 3510 | 2 | −6.3 | 0 | 0 |
| 2e | 391.8 | 50.02 | 100 | 6.16 | 5.3 | 17.4 | 0.20 | −6.6 | 0.78 | 666.0 | 183.0 | 45.6 | 288.3 | 149.0 | 1146.6 | 39.9 | 3659 | 3 | −6.2 | 1 | 1 |
| 2d | 357.3 | 52.10 | 100 | 6.01 | 4.8 | 16.5 | 0.04 | −5.8 | 0.63 | 632.7 | 183.5 | 45.8 | 325.5 | 77.8 | 1090.5 | 38.0 | 3641 | 4 | −6.1 | 1 | 0 |
| 2c | 353.4 | 35.7 | 100 | 5.70 | 5.8 | 16.9 | 0.08 | −6.4 | 1.14 | 660.2 | 192.8 | 47.5 | 342.0 | 77.8 | 1157.7 | 41.3 | 3508 | 1 | −6.3 | 1 | 1 |
| 2b | 387.4 | 58.9 | 100 | 6.00 | 4.9 | 17.7 | −0.02 | −6.2 | 0.67 | 674.2 | 266.2 | 45.4 | 284.5 | 77.9 | 1177.9 | 40.4 | 3669 | 4 | −6.1 | 1 | 0 |
| Rec. Values | 130–725 | 7–200 | >80% high <25% low | 1.0–12.5 | -2.0–6.5 | 8.0–35 | -3–1.2 | -6.0–0.5 | -1.5–1.5 | 300–1000 | 0.0–750.0 | 7.0–330.0 | 0.0–450.0 | 0.0–175.0 | 500–2000 | 13.0–70.0 | <25 poor >500 great | 1–8 | Below −5 | <3 | <4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawash, M.; Qaoud, M.T.; Jaradat, N.; Abdallah, S.; Issa, S.; Adnan, N.; Hoshya, M.; Sobuh, S.; Hawash, Z. Anticancer Activity of Thiophene Carboxamide Derivatives as CA-4 Biomimetics: Synthesis, Biological Potency, 3D Spheroid Model, and Molecular Dynamics Simulation. Biomimetics 2022, 7, 247. https://doi.org/10.3390/biomimetics7040247
Hawash M, Qaoud MT, Jaradat N, Abdallah S, Issa S, Adnan N, Hoshya M, Sobuh S, Hawash Z. Anticancer Activity of Thiophene Carboxamide Derivatives as CA-4 Biomimetics: Synthesis, Biological Potency, 3D Spheroid Model, and Molecular Dynamics Simulation. Biomimetics. 2022; 7(4):247. https://doi.org/10.3390/biomimetics7040247
Chicago/Turabian StyleHawash, Mohammed, Mohammed T. Qaoud, Nidal Jaradat, Samer Abdallah, Shahd Issa, Nawal Adnan, Marah Hoshya, Shorooq Sobuh, and Zafer Hawash. 2022. "Anticancer Activity of Thiophene Carboxamide Derivatives as CA-4 Biomimetics: Synthesis, Biological Potency, 3D Spheroid Model, and Molecular Dynamics Simulation" Biomimetics 7, no. 4: 247. https://doi.org/10.3390/biomimetics7040247