
Probing Small-Angle Molecular Motions with EPR Spectroscopy: Dynamical Transition and Molecular Packing in Disordered Solids


Abstract
:1. Introduction
2. Methodology
2.1. CW EPR Spectra of Nitroxides in Molecular Glasses
2.2. CW EPR: Dynamical Librations
2.3. ESE: Stochastic Librations
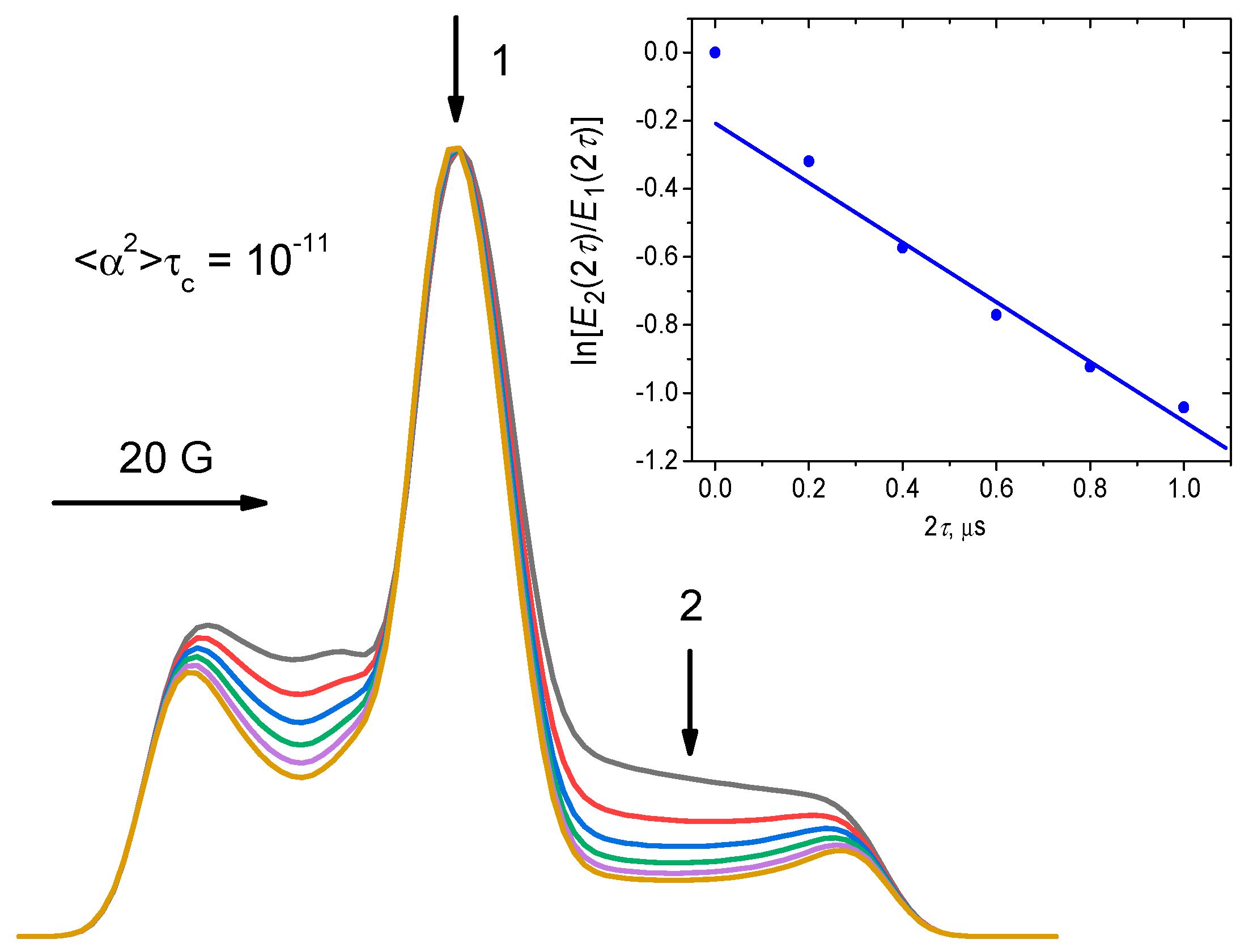
2.4. Stimulated ESE: Slow Motions
3. General Features of Small-Angle Motions in Solids
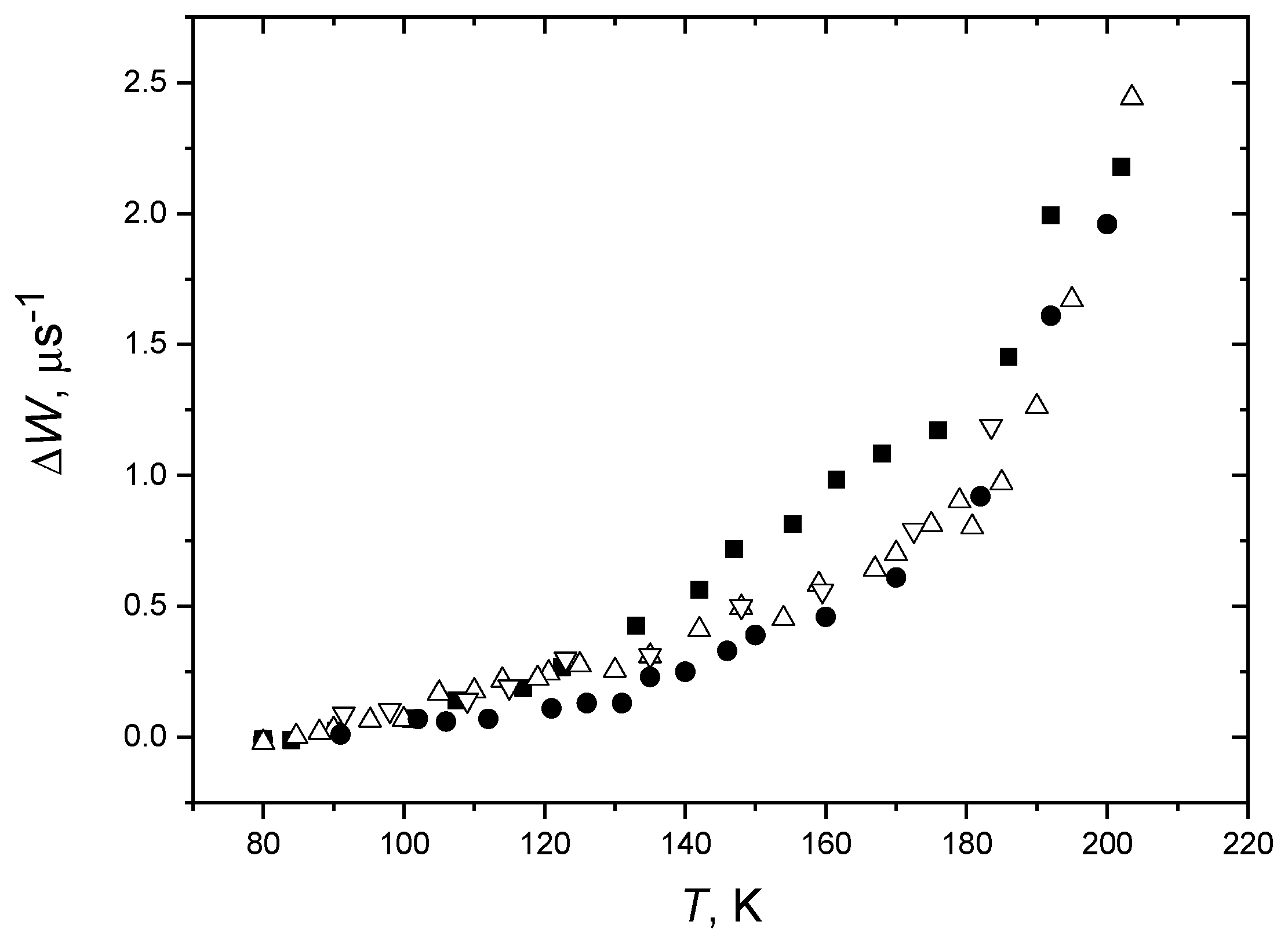
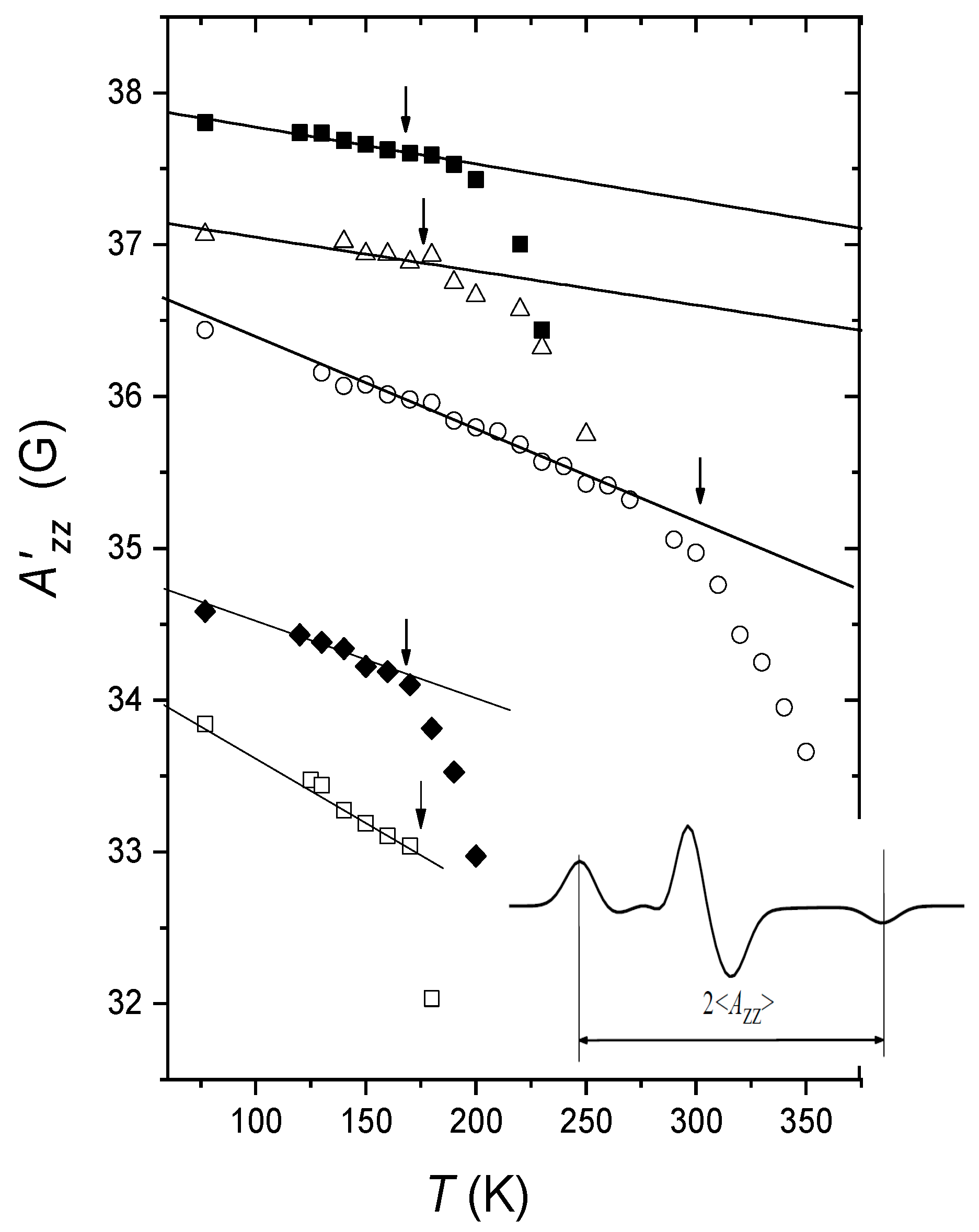
3.1. Dynamical Librations and Transition in Molecular Glasses
3.2. Cooperativity of Stochastic Librations, Influence of Hydration for Biological Systems
3.3. Individual Stochastic Librations on an Inorganic Surface
3.4. Stochastic Librations and Softness/Rigidity of Molecular Packing
4. Applications
4.1. Dynamical Transition in Membranes and Proteins
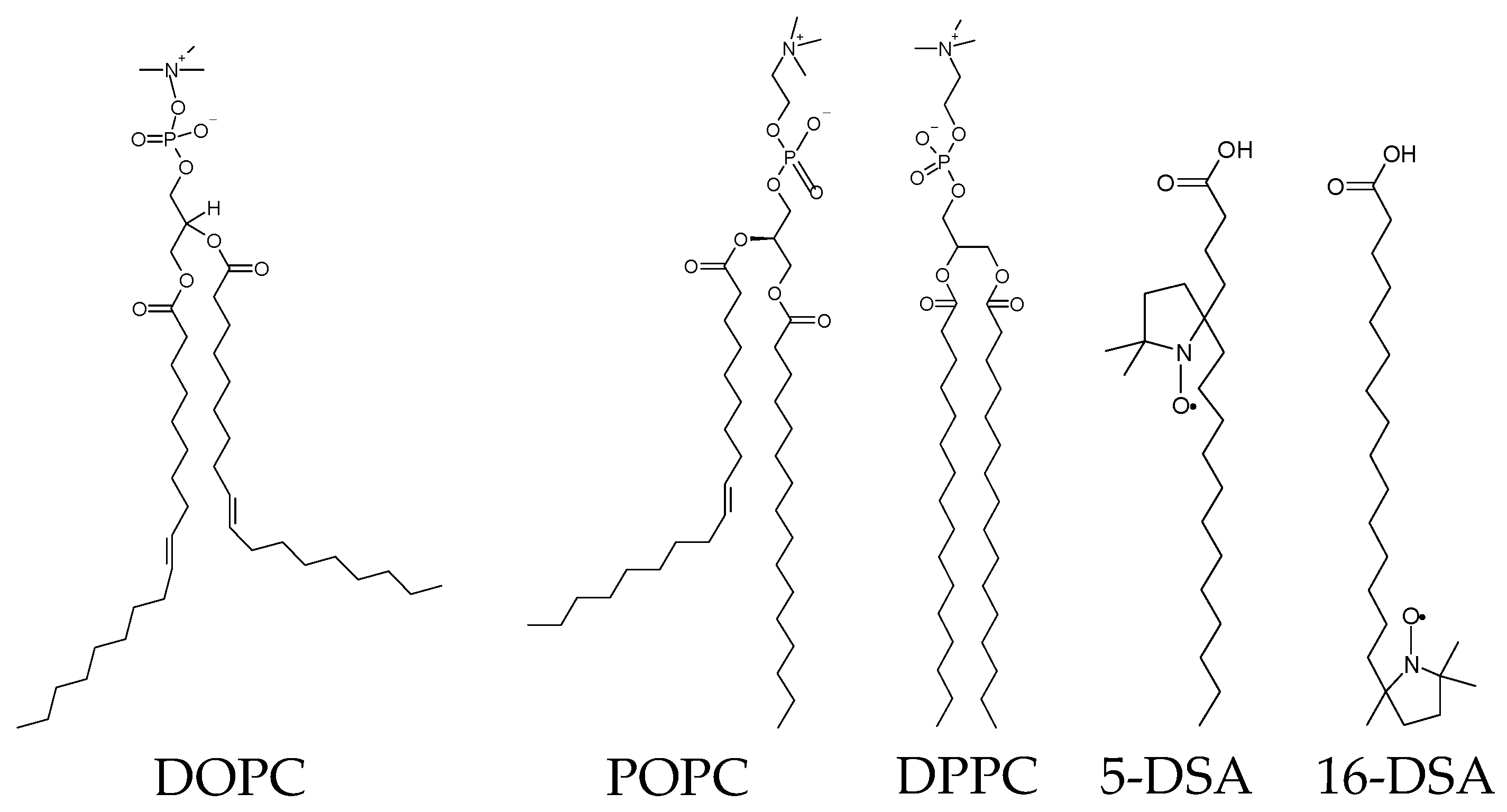
4.2. Lipid Packing in Biological Membranes
4.3. Stochastic Librations and Slow Rotations near Td in Membranes
4.4. Proteins and Antimicrobial Peptides in Membranes
- Ac-TOAC-Pro-Aib-Ala-Aib-Ala-Glu(OMe)-Aib-Val-Aib-Gly-Leu-Aib-Pro-Val-Aib-Aib-Glu(OMe)-Glu(OMe)-Phol [TOAC1]
- Ac-Aib-Pro-Aib-Ala-Aib-Ala-Glu(OMe)-TOAC-Val-Aib-Gly-Leu-Aib-Pro-Val-Aib-Aib-Glu(OMe)-Glu(OMe)-Phol [TOAC8]
- Ac-Aib-Pro-Aib-Ala-Aib-Ala-Glu(OMe)-Aib-Val-Aib-Gly-Leu-Aib-Pro-Val-TOAC-Aib-Glu(OMe)-Glu(OMe)-Phol [TOAC16].
- nOct-Aib1-Gly-Leu-Aib-Gly-Gly-Leu-Aib8-Gly-Ile-Lol (native trichogin GA IV)
- Fmoc-TOAC1-Gly-Leu-Aib4-Gly-Gly-Leu-Aib8-Gly-Ile-Leu-OMe (FTOAC1)
- Fmoc-Aib1-Gly-Leu-Aib4-Gly-Gly-Leu-TOAC8-Gly-Ile-Leu-OMe (FTOAC8).
4.5. Lipid Bilayers Interacting with Cryoprotectants
4.6. Supercooled Ionic Liquids
4.7. Supercooled Deep Eutectic Solvents
4.8. Intrinsically Disordered Proteins
4.9. Molecular Glasses and Other Systems
4.10. NMR of Small-Angle Motions, Secondary Relaxation
5. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paul, W.; Smith, G.D. Structure and dynamics of amorphous polymers: Computer simulations comared to experiment and theory. Rep. Prog. Phys. 2004, 67, 1117–1185. [Google Scholar] [CrossRef]
- Saragi, T.P.I.; Spehr, T.; Siebert, A.; Fuhrmann-Lieker, T.; Salbeck, J. Spiro compounds for organic optoelectronics. Chem. Rev. 2007, 107, 1011–1065. [Google Scholar] [CrossRef] [PubMed]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [Green Version]
- Zeller, R.C.; Pohl, R.O. Thermal conductivity and specific heat of noncrystalline solids. Phys. Rev. B 1971, 4, 2029. [Google Scholar] [CrossRef]
- Hunklinger, S.; Arnold, W. Ultrasonic Properties of Glasses at Low Temperatures; Academic Press: Cambridge, MA, USA, 1976; pp. 155–215. [Google Scholar]
- Pohl, R.O.; Liu, X.; Thompson, E. Low-temperature thermal conductivity and acoustic attenuation in amorphous solids. Rev. Mod. Phys. 2002, 74, 991–1013. [Google Scholar] [CrossRef]
- Phillips, W.A. Tunneling states in amorphous solids. J. Low Temp. Phys. 1972, 7, 351–360. [Google Scholar] [CrossRef]
- Anderson, P.W.; Halperin, B.I.; Varma, C.M. Anomalous low-temperature thermal properties of glasses and spin glasses. Philos. Mag. 1972, 25, 1–9. [Google Scholar] [CrossRef]
- Phillips, W.A. Amorphous Solids: Low-Temperature Properties. In Current Physics 24; Springer: Berlin/Heidelberg, Germany, 1981. [Google Scholar]
- Buchenau, U.; Nücker, N.; Dianoux, A.J. Neutron scattering study of the low-frequency vibrations in vitreous silica. Phys. Rev. Lett. 1984, 53, 2316–2319. [Google Scholar] [CrossRef]
- Malinovsky, V.K.; Novikov, V.N.; Sokolov, A.P. Log-normal spectrum of low-energy vibrational excitations in glasses. Phys. Lett. A 1991, 153, 63–66. [Google Scholar] [CrossRef]
- Lubchenko, V. Low-temperature anomalies in disordered solids: A cold case of contested relics? Adv. Phys. X 2018, 3, 1510296. [Google Scholar] [CrossRef] [Green Version]
- Lerner, E.; Bouchbinder, E. Low-energy quasilocalized excitations in structural glasses. J. Chem. Phys. 2021, 155, 200901. [Google Scholar] [CrossRef]
- Karpov, V.G.; Klinger, M.I.; Ignatiev, P.N. Atomic tunneling states and low-temperature anomalies of thermal properties in amorphous materials. Solid State Commun. 1982, 44, 333–337. [Google Scholar] [CrossRef]
- Galperin, Y.M.; Karpov, V.G.; Kozub, V.I. Localized states in glasses. Adv. Phys. 1989, 38, 669–737. [Google Scholar] [CrossRef]
- Buchenau, U. Soft localized vibrations in glasses and undercooled liquids. Philos. Mag. B 1992, 65, 303–315. [Google Scholar] [CrossRef]
- Gurevich, V.L.; Parshin, D.A.; Pelous, J.; Schober, H.R. Theory of low energy Raman scattering in glasses. Phys. Rev. B 1993, 48, 16318–16331. [Google Scholar] [CrossRef]
- Khodadadi, S.; Sokolov, A.P. Protein dynamics: From rattling in a cage to structural relaxation. Soft Matter. 2015, 11, 4984–4998. [Google Scholar] [CrossRef] [Green Version]
- Leporini, D.; Schädler, V.; Wiesner, U.; Spiess, H.W.; Jeschke, G. Electron spin relaxation due to small-angle motion: Theory for the canonical orientations and application to hierarchic cage dynamics in ionomers. J. Chem. Phys. 2003, 119, 11829–11846. [Google Scholar] [CrossRef]
- Bogdanov, A.V.; Vorobiev, A.K. Orientation order and rotation mobility of nitroxide biradicals determined by quantitative simulation of EPR spectra. Phys. Chem. Chem. Phys. 2016, 18, 31144–31153. [Google Scholar] [CrossRef]
- Vorobiev, A.K.; Bogdanov, A.V.; Yankova, T.S.; Chumakova, N.A. Spin probe determination of molecular orientation distribution and rotational mobility in liquid crystals: Model-free approach. J. Phys. Chem. B 2019, 123, 5875–5891. [Google Scholar] [CrossRef]
- Johari, G.P.; Goldstein, M.J. Viscous liquids and the glass transition. II. Secondary relaxations in glasses of rigid molecules. Chem. Phys. 1970, 53, 2372–2389. [Google Scholar] [CrossRef]
- Johari, G.P. Intrinsic mobility of molecular glasses. J. Chem. Phys. 1973, 58, 1766–1771. [Google Scholar] [CrossRef]
- Ngai, K.L.; Paluch, M. Classification of secondary relaxation in glass-formers based on dynamic properties. J. Chem. Phys. 2004, 120, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Gainaru, C.; Böhmer, R.; Kahlau, R.; Rössler, E. Energy landscape in molecular glasses probed by high-resolution dielectric experiments. Phys. Rev. B 2010, 82, 104205. [Google Scholar] [CrossRef]
- Parak, F.G. Proteins in action: The physics of structural fluctuations and conformational changes. Curr. Opin. Struct. Biol. 2003, 13, 552–557. [Google Scholar] [CrossRef]
- Doster, W. The protein-solvent glass transition. Biochim. Biophys. Acta 2010, 1804, 3–14. [Google Scholar] [CrossRef]
- Schiró, G. Anharmonic onsets in polypeptides revealed by neutron scattering: Experimental evidences and quantitative description of energy resolution dependence. Biophys. Chem. 2013, 180, 29–36. [Google Scholar] [CrossRef]
- Böhmer, R.; Chamberlin, R.V.; Diezemann, G.; Geil, B.; Heuer, A.; Hinze, G.; Kuebler, S.C.; Richert, R.; Schiener, B.; Sillescu, H.; et al. Nature of the non-exponential primary relaxation in structural glass-formers probed by dynamically selective experiments. J. Non-Cryst. Solids 1998, 235, 1–9. [Google Scholar] [CrossRef]
- Ediger, M.D. Spatially heterogeneous dynamics in supercooled liquids. Annu. Rev. Phys. Chem. 2000, 51, 99–128. [Google Scholar] [CrossRef] [Green Version]
- Vogel, M.; Rössler, E.A. Slow β process in simple organic glass formers studied by one- and two-dimensional 2H nuclear magnetic resonance. I. J. Chem. Phys. 2001, 114, 5802. [Google Scholar] [CrossRef]
- Vogel, M.; Medick, P.; Rössler, E.A. Secondary relaxation processes in molecular glasses studied by nuclear magnetic resonance spectroscopy. Annu. Rep. NMR Spectrosc. 2005, 56, 231–299. [Google Scholar] [CrossRef]
- Götze, W. Recent tests of the mode-coupling theory for glassy dynamics. J. Phys. Condens. Matter 1999, 11, A1–A45. [Google Scholar] [CrossRef]
- Ishii, K.; Nakayama, H. Structural relaxation of vapor-deposited molecular glasses and supercooled liquids. Phys. Chem. Chem. Phys. 2014, 16, 12073–12092. [Google Scholar] [CrossRef]
- Novikov, V.N.; Sokolov, A.P.; Strube, B.; Surovtsev, N.V.; Duval, E.; Mermet, A. Connection between quasielastic Raman scattering and free volume in polymeric glasses and supercooled liquids. J. Chem. Phys. 1997, 107, 1057. [Google Scholar] [CrossRef]
- Goldman, S.A.; Bruno, G.V.; Polnaszek, C.F.; Freed, J.H. An ESR study of anisotropic rotational reorientation and slow tumbling in liquid and frozen media. J. Chem. Phys. 1972, 56, 716. [Google Scholar] [CrossRef]
- Millhauser, G.L.; Freed, J.H. Two-dimensional electron spin echo spectroscopy and slow motions. J. Chem. Phys. 1984, 81, 37–48. [Google Scholar] [CrossRef]
- Dzuba, S.A.; Maryasov, A.G.; Salikhov, K.M.; Tsvetkov, Y.D. Superslow rotations of nitroxide radicals studied by pulse EPR spectroscopy. J. Magn. Reson. 1984, 58, 95–117. [Google Scholar] [CrossRef]
- Saxena, S.; Freed, J.H. Two-dimensional electron spin resonance and slow motions. J. Phys. Chem. A 1997, 101, 7998–8008. [Google Scholar] [CrossRef]
- Marsh, D. Spin-Label Electron Paramagnetic Resonance Spectroscopy; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar] [CrossRef]
- Van, S.P.; Birrell, G.B.; Griffith, O.H. Rapid anisotropic motion of spin labels. Models for motion averaging of the ESR parameters. J. Magn. Reson. 1974, 15, 444–459. [Google Scholar] [CrossRef]
- Kulik, L.V.; Rapatsky, L.L.; Pivtsov, A.V.; Surovtsev, N.V.; Adichtchev, S.V.; Grigor’ev, I.A.; Dzuba, S.A. Electron-nuclear double resonance study of molecular librations of nitroxides in molecular glasses: Quantum effects at low temperatures, comparison with low-frequency Raman scattering. J. Chem. Phys. 2009, 131, 064505. [Google Scholar] [CrossRef]
- Paschenko, S.V.; Toropov, Y.V.; Dzuba, S.A.; Tsvetkov, Y.D.; Vorobiev, A.K. Temperature dependence of amplitudes of libration motion of guest spin probe molecules in organic glasses. J. Chem. Phys. 1999, 110, 8150–8154. [Google Scholar] [CrossRef]
- Angell, C.A. Relaxation in liquids, polymers and plastic crystals—Strong fragile patterns and problems. J. Non-Cryst. Solids 1991, 131, 13–31. [Google Scholar] [CrossRef]
- Pivtsov, A.V.; Kulik, L.V.; Surovtsev, N.V.; Adichtchev, S.V.; Kirilyuk, I.A.; Grigor’ev, I.A.; Fedin, M.V.; Dzuba, S.A. Temperature dependence of hyperfine interaction for a 15N-nitroxide in a glassy matrix at 10–210 K. Appl. Magn. Reson. 2011, 41, 411–429. [Google Scholar] [CrossRef]
- Dzuba, S.A.; Tsvetkov, Y.D.; Maryasov, A.G. Echo-induced EPR spectra of nitroxides in organic glasses: Model of molecular orientational motion near equilibrium position. Chem. Phys. Lett. 1992, 188, 217–222. [Google Scholar] [CrossRef]
- Dzuba, S.A. Echo-induced EPR spectra of nitroxides: Study of molecular librations. Pure Appl. Chem. 1992, 64, 825–831. [Google Scholar] [CrossRef]
- Kirilina, E.P.; Dzuba, S.A.; Maryasov, A.G.; Tsvetkov, Y.D. Librational dynamics of nitroxide molecules in a molecular glass studied by echo-detected EPR. Appl. Magn. Reson. 2001, 21, 203–221. [Google Scholar] [CrossRef]
- Klauder, J.R.; Anderson, P.W. Spectral diffusion decay in spin resonance experiments. Phys. Rev. 1962, 125, 912–932. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Clarendon Press: Oxford, UK, 1961. [Google Scholar]
- Dzuba, S.A. Librational motion of guest spin probe molecules in glassy media. Phys. Lett. A 1996, 213, 77–84. [Google Scholar] [CrossRef]
- Dzuba, S.A. Librational motion of guest spin probe molecules in organic glasses: CW EPR and electron spin echo study. Spectrochim. Acta A 2000, 56, 227–234. [Google Scholar] [CrossRef]
- Dzuba, S.A.; Tsvetkov, Y.D. Magnetization transfer in pulsed EPR of N nitroxides: Reorientational motion model of molecules in glassy liquids. Chem. Phys. 1988, 120, 291–298. [Google Scholar] [CrossRef]
- Isaev, N.P.; Dzuba, S.A. Nitrogen nuclear spin flips in nitroxide spin probes of different sizes in glassy o-terphenyl: Possible relation with α- and β-relaxations. J. Chem. Phys. 2011, 135, 094508. [Google Scholar] [CrossRef] [PubMed]
- Syryamina, V.N.; Dzuba, S.A. Phospholipid bilayer relaxation dynamics as revealed by the pulsed electron-electron double resonance of spin labels. J. Chem. Phys. 2012, 137, 145102. [Google Scholar] [CrossRef] [PubMed]
- Du, J.L.; Eaton, G.R.; Eaton, S.S. Temperature, orientation, and solvent dependence of electron spin-lattice relaxation rates for nitroxyl radicals in glassy solvents and doped solid. J. Magn. Reason. A 1995, 115, 213–221. [Google Scholar] [CrossRef]
- Sato, H.; Kathirvelu, V.; Spagnol, G.; Rajca, S.; Rajca, A.; Eaton, S.S.; Eaton, G.R. Impact of electron-electron spin interaction on electron spin relaxation of nitroxide diradicals and tetraradical in glassy solvents between 10 and 300 K. J. Phys. Chem. B 2008, 112, 2818–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Bottle, S.E.; Blinco, J.P.; Micallef, A.S.; Eaton, G.R.; Eaton, S.S. Electron spin-lattice relaxation of nitroxyl radicals in temperature ranges that span glassy solutions to low-viscosity liquids. J. Magn. Reson. 2008, 191, 66–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sannikova, N.; Timofeev, I.; Bagryanskaya, E.; Bowman, M.; Fedin, M.; Krumkacheva, O. Electron spin relaxation of photoexcited porphyrin in water-glycerol glass. Molecules 2020, 25, 2677. [Google Scholar] [CrossRef]
- Kveder, M.; Merunka, D.; Jokic, M.; Makarevic, J.; Rakvin, B. Electron spin-lattice relaxation in solid ethanol: Effect of nitroxyl radical hydrogen bonding and matrix disorder. Phys. Rev. B 2009, 80, 052201. [Google Scholar] [CrossRef] [Green Version]
- Erilov, D.A.; Bartucci, R.; Guzzi, R.; Marsh, D.; Dzuba, S.A.; Sportelli, L. Echo-detected electron paramagnetic resonance spectra of spin-labeled lipids in membrane model systems. J. Phys. Chem. B 2004, 108, 4501–4507. [Google Scholar] [CrossRef] [Green Version]
- Isaev, N.P.; Fedin, M.V.; Dzuba, S.A. X- and Q-band electron spin echo study of stochastic molecular librations of spin labels in lipid bilayers. Appl. Magn. Reson. 2013, 44, 133–142. [Google Scholar] [CrossRef]
- Kirilina, E.P.; Prisner, T.F.; Bennati, M.; Endeward, B.; Dzuba, S.A.; Fuchs, M.R.; Möbius, K.; Schnegg, A. Molecular dynamics of nitroxides in glasses as studied by multifrequency EPR. Magn. Reson. Chem. 2005, 43, S119–S129. [Google Scholar] [CrossRef]
- Savitsky, A.; Plato, M.; Möbius, K. The temperature dependence of nitroxide spin–label interaction parameters: A high-field EPR study of intramolecular motional contributions. Appl. Magn. Reson. 2010, 37, 415–434. [Google Scholar] [CrossRef] [Green Version]
- Hertel, M.M.; Denysenkov, V.P.; Bennati, M.; Prisner, T.F. Pulsed 180-GHz EPR/ENDOR/PELDOR spectroscopy. Magn. Reson. Chem. 2005, 43, S248–S255. [Google Scholar] [CrossRef]
- Rohrer, M.; Gast, P.; Möbius, K.; Prisner, T.F. Anisotropic motion of semiquinones in photosynthetic reaction centers of Rhodobacter sphaeroides R26 and in frozen isopropanol solution as measured by pulsed high-field EPR at 95 GHz. Chem. Phys. Lett. 1996, 259, 523–530. [Google Scholar] [CrossRef]
- Schnegg, A.; Fuhs, M.; Rohrer, M.; Lubitz, W.; Prisner, T.F.; Möbius, K. Molecular dynamics of QA- and QB- in photosynthetic bacterial reaction centers studied by pulsed high-field EPR at 95 GHz. J. Phys. Chem. B 2002, 106, 9454–9462. [Google Scholar] [CrossRef]
- Savitsky, A.; Möbius, K. High-field EPR. Photosynth. Res. 2009, 102, 311–333. [Google Scholar] [CrossRef]
- Steinhoff, H.-J.; Savitsky, A.; Wegener, C.; Pfeiffer, M.; Plato, M.; Möbius, K. High-field EPR studies of the structure and conformational changes of site-directed spin labeled bacteriorhodopsin. Biochim. Biophys. Acta 2000, 1457, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Bagryanskaya, E.G.; Polovyanenko, D.N.; Fedin, M.V.; Kulik, L.; Schnegg, A.; Savitsky, A.; Möbius, K.; Coleman, A.W.; Ananchenko, G.S.; Ripmeester, J.A. Multifrequency EPR study of the mobility of nitroxides in solid-state calixarene nanocapsules. Phys. Chem. Chem. Phys. 2009, 11, 6700–6707. [Google Scholar] [CrossRef]
- Kuzhelev, A.A.; Krumkacheva, O.A.; Ivanov, M.Y.; Prikhod’ko, S.A.; Adonin, N.Y.; Tormyshev, V.M.; Bowman, M.K.; Fedin, M.V.; Bagryanskaya, E.G. Pulse EPR of triarylmethyl probes: A new approach for the investigation of molecular motions in soft matter. J. Phys. Chem. B 2018, 122, 8624–8630. [Google Scholar] [CrossRef]
- Narr, E.; Zimmermann, H.; Godt, A.; Goldfarb, D.; Jeschke, G. Structure and dynamics of copper complexes with 2,2’:6’, 2’’-terpyridines in glassy matrices. Phys. Chem. Chem. Phys. 2003, 5, 3959–3967. [Google Scholar] [CrossRef]
- Barbon, A.; Bortolus, M.; Brustolon, M.; Comotti, A.; Maniero, A.L.; Segre, U.; Sozzani, P. Dynamics of the triplet state of a dithiophene in different solid matrixes studied by transient and pulse EPR techniques. J. Phys. Chem. B 2003, 107, 3325–3331. [Google Scholar] [CrossRef]
- Dzuba, S.A.; Kirilina, E.P.; Salnikov, E.S.; Kulik, L.V. Restricted orientational motion of nitroxides in molecular glasses: Direct estimation of the motional time scale basing on the comparative study of primary and stimulated electron spin echo decays. J. Chem. Phys. 2005, 122, 094702. [Google Scholar] [CrossRef] [PubMed]
- Isaev, N.P.; Dzuba, S.A. Fast stochastic librations and slow rotations of spin labeled stearic acids in a model phospholipid bilayer at cryogenic temperatures. J. Phys. Chem. B 2008, 112, 13285–13291. [Google Scholar] [CrossRef] [PubMed]
- Isaev, N.P.; Kulik, L.V.; Kirilyuk, I.A.; Reznikov, V.A.; Grigor’ev, I.A.; Dzuba, S.A. Fast stochastic librations and slow small-angle rotations of molecules in glasses observed on nitroxide spin probes by stimulated electron spin echo spectroscopy. J. Non-Cryst. Solids 2010, 356, 1037–1042. [Google Scholar] [CrossRef]
- Isaev, N.P.; Syryamina, V.N.; Dzuba, S.A. Small-angle orientational motions of spin labeled lipids in cholesterol-containing bilayers studied at low temperatures by electron spin echo spectroscopy. J. Phys. Chem. B 2010, 114, 9510–9515. [Google Scholar] [CrossRef] [PubMed]
- Syryamina, V.N.; Isaev, N.P.; Peggion, C.; Formaggio, F.; Toniolo, C.; Raap, J.; Dzuba, S.A. Small-amplitude backbone motions of the spin-labeled lipopeptide trichogin GA IV in a lipid membrane as revealed by electron spin echo. J. Phys. Chem. B 2010, 114, 12277–12283. [Google Scholar] [CrossRef]
- Ivanisenko, N.V.; Dzuba, S.A. Molecular motion in frozen phospholipid bilayers in the presence of sucrose and sorbitol studied by the spin-echo EPR of spin labels. Appl. Magn. Reson. 2013, 44, 883–891. [Google Scholar] [CrossRef]
- Valiev, K.A.; Ivanov, E.N. Rotational brownian motion. Sov. Phys. Uspekhi 1973, 16, 1. [Google Scholar] [CrossRef]
- Earle, K.A.; Budil, D.E.; Freed, J.H. 250-GHz EPR of nitroxides in the slow-motional regime: Models of rotational diffusion. J. Phys. Chem. 1993, 97, 13289–13297. [Google Scholar] [CrossRef]
- Nickels, J.D.; O’Neill, H.; Hong, L.; Tyagi, M.; Ehlers, G.; Weiss, K.L.; Zhang, Q.; Yi, Z.; Mamontov, E.; Smith, J.C.; et al. Dynamics of protein and its hydration water: Neutron scattering studies on fully deuterated GFP. Biophys. J. 2012, 103, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Nienhaus, G.U.; Frauenfelder, H.; Parak, F. Structural fluctuations in glass-forming liquids: Mössbauer spectroscopy on iron in glycerol. Phys. Rev. B 1991, 43, 3345–3350. [Google Scholar] [CrossRef]
- Wuttke, J.; Petry, W.; Coddens, G.; Fujara, F. Fast dynamics of glass-forming glycerol. Phys. Rev. E 1995, 52, 4026–4034. [Google Scholar] [CrossRef]
- Petry, W.; Bartsch, E.; Fujara, F.; Kiebel, M.; Sillescu, H.; Farago, B. Dynamic anomaly in the glass transition region of orthoterphenyl. Z. Phys. B Condens. Matter. 1991, 83, 175–184. [Google Scholar] [CrossRef]
- Tölle, A.; Zimmermann, H.; Fujara, F.; Petry, W.; Schmidt, W.; Schober, H.; Wuttke, J. Vibrational states of glassy and crystalline orthoterphenyl. Eur. Phys. J. B 2000, 16, 73–80. [Google Scholar] [CrossRef]
- Tölle, A. Neutron scattering studies of the model glass former ortho-terphenyl. Rep. Prog. Phys. 2001, 64, 1473–1532. [Google Scholar] [CrossRef] [Green Version]
- Plazanet, M.; Schober, H. Anharmonicity in a fragile glass-former probed by inelastic neutron scattering. Phys. Chem. Chem. Phys. 2008, 10, 5723–5729. [Google Scholar] [CrossRef]
- Surovtsev, N.V.; Malinovsky, V.K.; Boldyreva, E.V. Raman study of low-frequency modes in three glycine polymorphs. J. Chem. Phys. 2011, 134, 045102. [Google Scholar] [CrossRef]
- Roh, J.H.; Briber, R.M.; Damjanovic, A.; Thirumalai, D.; Woodson, S.A.; Sokolov, A.P. Dynamics of tRNA at different levels of hydration. Biophys. J. 2009, 96, 2755–2762. [Google Scholar] [CrossRef] [Green Version]
- Fitter, J.; Lechner, R.E.; Dencher, N.A. Interactions of hydration water and biological membranes studied by neutron scattering. J. Phys. Chem. 1999, 103, 8036–8050. [Google Scholar] [CrossRef]
- Wood, K.; Plazanet, M.; Gabel, F.; Kessler, B.; Oesterhelt, D.; Zaccai, G.; Weik, M. Dynamics of hydration water in deuterated purple membranes explored by neutron scattering. Eur. Biophys. J. 2008, 37, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Tobias, D.J.; Senguptaa, N.; Tarek, M. Hydration dynamics of purple membranes. Farad. Discuss. 2009, 141, 99–116. [Google Scholar] [CrossRef]
- Frölich, A.; Gabel, F.; Jasnin, M.; Lehnert, U.; Oesterhelt, D.; Stadler, A.; Tehei, M.; Weik, M.; Wood, K.; Zaccai, G. From shell to cell: Neutron scattering studies of biological water dynamics and coupling to activity. Farad. Discuss. 2009, 141, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Mamontov, E.; Sharma, V.K.; Borreguero, J.M.; Tyagi, M. Protein-style dynamical transition in a non-biological polymer and a non-aqueous solvent. J. Phys. Chem. B 2016, 120, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Liu, L.; Fratini, E.; Baglioni, P.; Faraone, A.; Mamontov, E. Observation of fragile-to-strong dynamic crossover in protein hydration water. Proc. Natl. Acad. Sci. USA 2006, 103, 9012–9016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetto, A.; Kearley, G.J. Elastic scattering spectroscopy (ESS): An instrument-concept for dynamics of complex (bio-) systems from elastic neutron scattering. Sci. Rep. 2016, 6, 34266. [Google Scholar] [CrossRef] [PubMed]
- Magazù, S.; Mezei, F.; Falus, P.; Farago, B.; Mamontov, E.; Russina, M.; Migliardo, F. Protein dynamics as seen by (quasi) elastic neutron scattering. Biochim. Biophys. Acta B 2017, 1861, 3504–3512. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Fenimore, P.W.; Frauenfelder, H.; Mezei, F.; Swenson, J.; Young, R.D. Protein fluctuations explored by inelastic neutron scattering and dielectric relaxation spectroscopy. Philos. Mag. 2008, 88, 3877–3883. [Google Scholar] [CrossRef]
- Khodadadi, S.; Pawlus, S.; Roh, J.H.; Sakai, V.G.; Mamontov, E.; Sokolov, A.P. The origin of the dynamical transition in proteins. J. Chem. Phys. 2008, 128, 195106. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Hayward, J.A.; Finney, J.L.; Daniel, R.M.; Smith, J.C. Neutron frequency windows and the protein dynamical transition. Biophys. J. 2004, 87, 1436–1444. [Google Scholar] [CrossRef] [Green Version]
- Dzuba, S.A.; Salnikov, E.S.; Kulik, L.V. CW EPR, echo-detected EPR, and field-step ELDOR study of molecular motions of nitroxides in o-terphenyl glass: Dynamical transition, dynamical heterogeneity and β relaxation. Appl. Magn. Reson. 2006, 30, 637–650. [Google Scholar] [CrossRef]
- Golysheva, E.A.; Shevelev, G.Y.; Dzuba, S.A. Dynamical transition in molecular glasses and proteins observed by spin relaxation of nitroxide spin probes and labels. J. Chem. Phys. 2017, 147, 064501. [Google Scholar] [CrossRef]
- Kirilina, E.P.; Grigoriev, I.A.; Dzuba, S.A. Orientational motion of nitroxides in molecular glasses: Dependence on the chemical structure, on the molecular size of the probe, and on the type of the matrix. J. Chem. Phys. 2004, 121, 12465–12471. [Google Scholar] [CrossRef]
- Golysheva, E.A.; de Zotti, M.; Toniolo, C.; Formaggio, F.; Dzuba, S.A. Low-temperature dynamical transition in lipid bilayers detected by spin-label ESE spectroscopy. Appl. Magn. Reson. 2018, 49, 1369–1383. [Google Scholar] [CrossRef]
- Scarpelli, F.; Bartucci, R.; Sportelli, L.; Guzzi, R. Solvent effect on librational dynamics of spin-labelled haemoglobin by ED- and CW-EPR. Eur. Biophys, J. 2011, 40, 273–279. [Google Scholar] [CrossRef]
- Maslennikova, N.A.; Golysheva, E.A.; Dzuba, S.A. Evidence for an ordering transition near 120 K in an intrinsically disordered protein, casein. Molecules 2021, 26, 5971. [Google Scholar] [CrossRef]
- Syryamina, V.N.; Dzuba, S.A. Dynamical transitions at low temperatures in the nearest hydration shell of phospholipid bilayers. J. Phys. Chem. B 2017, 121, 1026–1032. [Google Scholar] [CrossRef]
- Mamontov, E.; Chu, X.-Q. Water−protein dynamic coupling and new opportunities for probing it at low to physiological temperatures in aqueous solutions. Phys. Chem. Chem. Phys. 2012, 14, 11573–11588. [Google Scholar] [CrossRef]
- Golysheva, E.A.; Samoilova, R.I.; De Zotti, M.; Toniolo, C.; Formaggio, F.; Dzuba, S.A. Electron spin echo detection of stochastic molecular librations: Non-cooperative motions on solid surface. J. Magn. Reson. 2019, 309, 106621. [Google Scholar] [CrossRef]
- Golysheva, E.A.; Samoilova, R.I.; De Zotti, M.; Formaggio, F.; Gobbo, M.; Dzuba, S.A. ESE-detected molecular motions of spin-labeled molecules on a solid inorganic surface: Motional models and onset temperatures. Appl. Magn. Reson. 2020, 51, 1019–1029. [Google Scholar] [CrossRef]
- Salikhov, K.M.; Dzuba, S.A.; Raitsimring, A.M. The theory of electron spin echo signal decay resulting from dipole-dipole interactions between paramagnetic centers in solids. J. Magn. Reson. 1981, 42, 255–276. [Google Scholar] [CrossRef]
- Aloi, E.; Guzzi, R.; Bartucci, R. Unsaturated lipid bilayers at cryogenic temperature: Librational dynamics of chain-labeled lipids from pulsed and CW-EPR. Phys. Chem. Chem. Phys. 2019, 21, 18699–18705. [Google Scholar] [CrossRef]
- Surovtsev, N.V.; Ivanisenko, N.V.; Kirillov, K.Y.; Dzuba, S.A. Low-temperature dynamical and structural properties of saturated and monounsaturated phospholipid bilayers revealed by Raman and spin-label EPR spectroscopy. J. Phys. Chem. B 2012, 116, 8139–8144. [Google Scholar] [CrossRef] [PubMed]
- Konov, K.B.; Isaev, N.P.; Dzuba, S.A. Low-temperature molecular motions in phospholipid bilayers in presence of glycerol as studied by spin-echo EPR of spin labels. Appl. Magn. Reson. 2014, 45, 1117–1126. [Google Scholar] [CrossRef]
- Konov, K.B.; Isaev, N.P.; Dzuba, S.A. Low-temperature molecular motions in lipid bilayers in the presence of sugars: Insights into cryoprotective mechanisms. J. Phys. Chem. B 2014, 118, 12478–12485. [Google Scholar] [CrossRef] [PubMed]
- Surovtsev, N.V.; Dzuba, S.A. Conformational changes of lipids in bilayers at the dynamical transition near 200 K seen by Raman scattering. J. Phys. Chem. B 2009, 113, 15558–15562. [Google Scholar] [CrossRef]
- Isaev, N.P.; Syryamina, V.N.; Dzuba, S.A. Influence of cholesterol on molecular motions in spin-labeled lipid bilayers observed by stimulated ESE. Appl. Magn. Reson. 2010, 37, 405–413. [Google Scholar] [CrossRef]
- Erilov, D.A.; Bartucci, R.; Guzzi, R.; Marsh, D.; Dzuba, S.A.; Sportelli, L. Librational motion of spin-labeled lipids in high-cholesterol containing membranes from echo-detected EPR spectra. Biophys. J. 2004, 87, 3873–3881. [Google Scholar] [CrossRef] [Green Version]
- Weik, M.; Lehnert, U.; Zaccai, G. Liquid-like water confined in stacks of biological membranes at 200 K and its relation to protein dynamics. Biophys. J. 2005, 89, 3639–3646. [Google Scholar] [CrossRef] [Green Version]
- Shalaev, E.Y.; Zografi, G.; Steponkus, P.L. Occurrence of glass transitions in long-chain phosphatidylcholine mesophases. J. Phys. Chem. B 2010, 114, 3526–3533. [Google Scholar] [CrossRef]
- De Simone, F.; Guzzi, R.; Sportelli, L.; Marsh, D.; Bartucci, R. Electron spin-echo studies of spin-labelled lipid membranes and free fatty acids interacting with human serum albumin. Biochim. Biophys. Acta 2007, 1768, 1541–1549. [Google Scholar] [CrossRef] [Green Version]
- Bartucci, R.; Guzzi, R.; De Zotti, M.; Toniolo, C.; Sportelli, L.; Marsh, D. Backbone dynamics of alamethicin bound to lipid membranes: Spin-echo electron paramagnetic resonance of TOAC-spin labels. Biophys. J. 2008, 94, 2698–2705. [Google Scholar] [CrossRef] [Green Version]
- Marsh, D.; Bartucci, R.; Guzzi, R.; Sportelli, L.; Esmann, M. Librational fluctuations in protein glasses. Biochim. Biophys. Acta 2013, 1834, 1591–1595. [Google Scholar] [CrossRef]
- Guzzi, R.; Bartucci, R.; Esmann, M.; Marsh, D. Lipid librations at the interface with the Na, K-ATPase. Biophys. J. 2015, 108, 2825–2832. [Google Scholar] [CrossRef] [Green Version]
- Aloi, E.; Oranges, M.; Guzzi, R.; Bartucci, R. Low-temperature dynamics of chain-labeled lipids in ester- and ether-linked phosphatidylcholine membranes. J. Phys. Chem. B 2017, 121, 9239–9246. [Google Scholar] [CrossRef]
- Aloi, E.; Bartucci, R. Interdigitated lamellar phases in the frozen state: Spin-label CW- and FT-EPR. Biophys. Chem. 2019, 253, 106229. [Google Scholar] [CrossRef]
- Aloi, E.; Bartucci, R. Influence of hydration on segmental chain librations and dynamical transition in lipid bilayers. Biochim. Biphys. Acta-Biomembr. 2022, 1864, 183805. [Google Scholar] [CrossRef]
- Ferrand, M.; Petry, W.; Dianoux, A.J.; Zaccai, G. Dynamical transition of bacteriorhodopsin in purple membranes revealed by neutron scattering: A relation between structure, dynamics and function. Phys. A 1993, 201, 425–429. [Google Scholar] [CrossRef]
- Wood, K.; Plazanet, M.; Gabel, F.; Kessler, B.; Oesterhelt, D.; Zaccai, G.; Weik, M. Coupling of protein and hydration-water dynamics in biological membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 18049–18054. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.; Marion, J.; Natali, F.; Kats, E.; Bicout, D.J. The dynamical transition of lipid multilamellar bilayers as a matter of cooperativity. J. Phys. Chem. B 2017, 121, 6860–6868. [Google Scholar] [CrossRef]
- Doxastakis, M.; Sakai, V.G.; Ohtake, S.; Maranas, J.K.; de Pablo, J.J. A molecular view of melting in anhydrous phospholipidic membranes. Biophys. J. 2007, 92, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Dzuba, S.A.; Kirilina, E.P.; Salnikov, E.S. On the possible manifestation of harmonic-anharmonic dynamical transition in glassy media in electron paramagnetic resonance of nitroxide spin probes. J. Chem. Phys. 2006, 125, 054502. [Google Scholar] [CrossRef]
- Golysheva, E.A.; Dzuba, S.A. Lipid chain mobility and packing in DOPC bilayers at cryogenic temperatures. Chem. Phys. Lipids 2020, 226, 104817. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, C.; Brückner, H. (Eds.) Peptaibiotics: Fungal Peptides Containing α-Dialkyl α-Amino Acids. In Verlag Helvetica Chimica Acta; Zurich and Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Duclohier, H. Antimicrobial peptides and peptaibols, substitutes for conventional antibiotics. Curr. Pharm. Des. 2010, 16, 3212–3223. [Google Scholar] [CrossRef] [PubMed]
- Bocchinfuso, G.; Palleschi, A.; Orioni, B.; Grande, G.; Formaggio, F.; Toniolo, C.; Park, Y.; Hahm, K.-S.; Stella, L. Different mechanisms of action of antimicrobial peptides: Insights from fluorescence spectroscopy experiments and molecular dynamics simulations. J. Pept. Sci. 2009, 15, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, C.; Crisma, M.; Formaggio, F. TOAC, a nitroxide spin-labeled, achiral C(alpha)-tetrasubstituted alpha-amino acid, is an excellent tool in material science and biochemistry. Pept. Sci. 1998, 47, 153–158. [Google Scholar] [CrossRef]
- Neumann, N.K.N.; Stoppacher, N.; Zeilinger, S.; Degenkolb, T.; Brückner, H.; Schuhmacher, R. The peptaibiotics database—A comprehensive online resource. Chem. Biodivers. 2015, 12, 743–751. [Google Scholar] [CrossRef]
- Crowe, J.H.; Carpenter, J.F.; Crowe, L.M. The role of vitrification in anhydrobiosis. Annu. Rev. Physiol. 1998, 60, 73–103. [Google Scholar] [CrossRef]
- Hoekstra, F.A.; Golovina, E.A.; Buitink, J. Mechanisms of plant desiccation tolerance. Trends Plant. Sci. 2001, 6, 431–438. [Google Scholar] [CrossRef]
- Leekumjorn, S.; Sum, A.K. Molecular investigation of the interactions of trehalose with lipid bilayers of DPPC, DPPE and their mixture. Mol. Simul. 2006, 32, 219–230. [Google Scholar] [CrossRef]
- Lairion, F.; Disalvo, E.A. Effect of trehalose on the contributions to the dipole potential of lipid monolayers. Chem. Phys. Lipids 2007, 150, 117–124. [Google Scholar] [CrossRef]
- Golovina, E.A.; Golovin, A.; Hoekstra, F.A.; Faller, R. Water replacement hypothesis in atomic details: Effect of trehalose on the structure of single dehydrated POPC bilayers. Langmuir 2010, 26, 11118–11126. [Google Scholar] [CrossRef]
- Koster, K.L.; Maddocks, K.J.; Bryant, G. Exclusion of maltodextrins from phosphatidylcholine multilayers during dehydration: Effects on membrane phase behavior. Eur. Biophys. J. 2003, 32, 96–105. [Google Scholar] [CrossRef]
- Lenné, T.; Garvey, C.J.; Koster, K.L.; Bryant, G. Effects of sugars on lipid bilayers during dehydration—SAXS/WAXS measurements and quantitative model. J. Phys. Chem. B 2009, 113, 2486–2491. [Google Scholar] [CrossRef]
- Kent, B.; Hunt, T.; Darwish, T.A.; Hauss, T.; Garvey, C.J.; Bryant, G. Localization of trehalose in partially hydrated DOPC bilayers: Insights into cryoprotective mechanisms. J. R. Soc. Interface 2014, 11, 20140069. [Google Scholar] [CrossRef]
- Westh, P. Glucose, sucrose and trehalose are partially excluded from the interface of hydrated DMPC bilayers. Phys. Chem. Chem. Phys. 2008, 10, 4110–4112. [Google Scholar] [CrossRef]
- Andersen, H.D.; Wang, C.; Arleth, L.; Peters, G.H.; Westh, P. Reconciliation of opposing views on membrane–sugar interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 1874–1878. [Google Scholar] [CrossRef] [Green Version]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and nanostructure in ionic liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [Green Version]
- Hallett, J.P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef]
- Rogers, R.D. Chemistry: Ionic liquids–solvents of the future? Science 2003, 302, 792–793. [Google Scholar] [CrossRef]
- Blanchard, L.A.; Hancu, D.; Beckman, E.J.; Brennecke, J.F. Green processing using ionic liquids and CO2. Nature 1999, 399, 28–29. [Google Scholar] [CrossRef]
- Egorova, K.S.; Gordeev, E.G.; Ananikov, V.P. Biological activity of ionic liquids and their application in pharmaceutics and medicine. Chem. Rev. 2017, 117, 7132–7189. [Google Scholar] [CrossRef]
- Watanabe, M.; Thomas, M.L.; Zhang, S.; Ueno, K.; Yasuda, T.; Dokko, K. Application of ionic liquids to energy storage and conversion materials and devices. Chem. Rev. 2017, 117, 7190–7239. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; Verma, R.D.; Meshri, D.T.; Shreeve, J.M. Energetic nitrogen-rich salts and ionic liquids. Angew. Chem. Int. Ed. 2006, 45, 3584–3601. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy applications of ionic liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef] [Green Version]
- Manthiram, A.; Fu, Y.; Chung, S.H.; Zu, C.; Su, Y.S. Rechargeable lithium-sulfur batteries. Chem. Rev. 2014, 114, 11751–11787. [Google Scholar] [CrossRef]
- van Rantwijk, F.; Sheldon, R.A. Biocatalysis in ionic liquids. Chem. Rev. 2007, 107, 2757–2785. [Google Scholar] [CrossRef] [PubMed]
- Giernoth, R. Task-specific ionic liquids. Angew. Chem. Int. Ed. 2010, 49, 2834–2839. [Google Scholar] [CrossRef] [PubMed]
- Wasserscheid, P.; Keim, W. Ionic liquids—New ‘solutions’ for transition metal catalysis. Angew. Chem. Int. Ed. 2000, 39, 3772–3789. [Google Scholar] [CrossRef]
- Trioli, A.; Russina, O.; Bleif, H.-J.; Di Cola, E. Nanoscale segregation in Room Temperature Ionic Liquids. J. Phys. Chem. B 2007, 111, 4641–4644. [Google Scholar] [CrossRef]
- Weingärtner, H. Understanding ionic liquids at the molecular level: Facts, problems, and controversies. Angew. Chem. Int. Ed. 2008, 47, 654–670. [Google Scholar] [CrossRef]
- Mogurampelly, S.; Keith, J.R.; Ganesan, V. Mechanisms underlying ion transport in polymerized ionic liquids. J. Am. Chem. Soc. 2017, 139, 9511–9514. [Google Scholar] [CrossRef]
- Ivanov, M.Y.; Krumkacheva, O.A.; Dzuba, S.A.; Fedin, M.V. Microscopic rigidity and heterogeneity of ionic liquids probed by stochastic molecular librations of the dissolved nitroxides. Phys. Chem. Chem. Phys. 2017, 19, 26158–26163. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, M.Y.; Prikhod’ko, S.A.; Adonin, N.Y.; Kirilyuk, I.A.; Adichtchev, S.V.; Surovtsev, N.V.; Dzuba, S.A.; Fedin, M.V. Structural anomalies in ionic liquids near the glass transition revealed by pulse EPR. J. Phys. Chem. Lett. 2018, 9, 4607–4612. [Google Scholar] [CrossRef]
- Ivanov, M.Y.; Fedin, M.V. Nanoscale heterogeneities in ionic liquids: Insights from EPR of spin probes. Mendeleev Commun. 2018, 28, 565–573. [Google Scholar] [CrossRef]
- Ivanov, M.Y.; Prikhod’ko, S.A.; Adonin, N.Y.; Fedin, M.V. Structural anomalies in binary mixtures of ionic liquid [Bmim]BF4 with water studied by EPR. J. Phys. Chem. B 2019, 123, 9956–9962. [Google Scholar] [CrossRef]
- Ivanov, M.Y.; Poryvaev, A.S.; Polyukhov, D.M.; Prikhod’ko, S.A.; Adonin, N.Y.; Fedin, M.V. Nanoconfinement effects on structural anomalies in imidazolium ionic liquids. Nanoscale 2020, 12, 23480–23487. [Google Scholar] [CrossRef]
- Bakulina, O.D.; Ivanov, M.Y.; Prikhod’ko, S.A.; Pylaeva, S.I.; Zaytseva, V.; Surovtsev, N.V.; Adonin, N.Y.; Fedin, M.V. Nanocage formation and structural anomalies in imidazolium ionic liquid glasses governed by alkyl chains of cations. Nanoscale 2020, 12, 19982–19991. [Google Scholar] [CrossRef]
- Ivanov, M.Y.; Bakulina, O.D.; Alimov, D.V.; Prikhod’ko, S.A.; Veber, S.L.; Pylaeva, S.; Adonin, N.Y.; Fedin, M.V. Inherent heterogeneities and nanostructural anomalies in organic glasses revealed by EPR. Nanoscale Adv. 2021, 3, 4973–4978. [Google Scholar] [CrossRef]
- Ivanov, M.Y.; Prikhod’ko, S.A.; Bakulina, O.D.; Kiryutin, A.S.; Adonin, N.Y.; Fedin, M.V. Validation of structural grounds for anomalous molecular mobility in ionic liquid glasses. Molecules 2021, 26, 5828. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Munro, H.L.; Rasheed, R.K.; Tambyrajah, V. Preparation of novel, moisture-stable, lewis-acidic ionic liquids containing quaternary ammonium salts with functional side chains. Chem. Commun. 2001, 19, 2010–2011. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep eutectic solvents (DESs) and their applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [Green Version]
- Paiva, A.; Craveiro, R.; Aroso, I.; Martins, M.; Reis, R.L.; Duarte, R.C. Natural deep eutectic solvents—Solvents for the 21st century. ACS Sustain. Chem. Eng. 2014, 2, 1063–1071. [Google Scholar] [CrossRef]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep eutectic solvents: A review of fundamentals and applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- Dai, Y.; Varypataki, E.M.; Golovina, E.A.; Jiskoot, W.; Witkamp, G.-J.; Choi, Y.H.; Verpoorte, R. Chapter Seven—Natural deep eutectic solvents in plants and plant cells: In vitro evidence for their possible functions. Adv. Botan. Res. 2021, 97, 159–184. [Google Scholar] [CrossRef]
- Reuter, D.; Munzner, P.; Gainaru, C.; Lunkenheimer, P.; Loidl, A.; Bohmer, R. Translational and reorientational dynamics in deep eutectic solvents. J. Chem. Phys. 2021, 154, 154501. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural deep eutectic solvents as new potential media for green technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, H.; Sharma, V.K.; Mukhopadhyay, R.; Mitra, S. Solvation and transport of lithium ions in deep eutectic solvents. J. Chem. Phys. 2020, 153, 104505. [Google Scholar] [CrossRef] [PubMed]
- Golysheva, E.A.; Dzuba, S.A. Low-temperature molecular motions in a deep eutectic solvent choline chloride/urea studied by spin-probe EPR. Russ. Chem. Bull. 2021, 70, 2366–2369. [Google Scholar] [CrossRef]
- Golysheva, E.A.; Maslennikova, N.A.; Baranov, D.S.; Dzuba, S.A. Structural properties of supercooled deep eutectic solvents: Choline chloride–thiourea compared to reline. Phys. Chem. Chem. Phys. 2022. [Google Scholar] [CrossRef]
- Mukesh, C.; Mondal, D.; Sharma, M.; Prasad, K. Choline chloride-thiourea, a deep eutectic solvent for the production of chitin nanofibers. Carbohydr. Polym. 2014, 103, 466–471. [Google Scholar] [CrossRef]
- Zainal-Abidin, M.H.; Hayyan, M.; Ngoh, G.C.; Wong, W.F.; Looi, C.Y. Emerging frontiers of deep eutectic solvents in drug discovery and drug delivery systems. J. Control. Release 2019, 316, 168–195. [Google Scholar] [CrossRef]
- Emami, S.; Shayanfar, A. Deep eutectic solvents for pharmaceutical formulation and drug delivery applications. Pharm. Dev. Technol. 2020, 25, 779–796. [Google Scholar] [CrossRef]
- Mbous, Y.P.; Hayyan, M.; Wong, W.F.; Hayyan, A.; Looi, C.Y.; Hashim, M.A. Simulation of deep eutectic solvents’ interaction with membranes of cancer cells using COSMO-RS. J. Phys. Chem. B 2020, 124, 9086–9094. [Google Scholar] [CrossRef]
- Wagle, D.V.; Zhao, H.; Baker, G.A. Deep eutectic solvents: Sustainable media for nanoscale and functional materials. Acc. Chem. Res. 2014, 47, 2299–2308. [Google Scholar] [CrossRef]
- Dunker, A.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.R.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Modell. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H. Intrinsically unstructured proteins: Reassessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta-Proteins Proteom. 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Deiana, A.; Forcelloni, S.; Porrello, A.; Giansanti, A. Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS ONE 2019, 14, e0217889. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.A.; Pettitt, B.M. Physical chemistry of the protein backbone: Enabling the mechanisms of intrinsic protein disorder. J. Phys. Chem. B 2020, 124, 4379–4390. [Google Scholar] [CrossRef]
- Hosoya, Y.; Ohkanda, J. Intrinsically disordered proteins as regulators of transient biological processes and as untapped drug targets. Molecules 2021, 26, 2118. [Google Scholar] [CrossRef]
- Uversky, V.N. Supramolecular fuzziness of intracellular liquid droplets: Liquid-liquid phase transitions, membrane-less organelles, and intrinsic disorder. Molecules 2019, 24, 3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milov, A.D.; Samoilova, R.I.; Shubin, A.A.; Grishin, Y.A.; Dzuba, S.A. ESEEM measurements of local water concentration in D2O-containing spin-labeled systems. Appl. Magn. Reson. 2008, 35, 73–94. [Google Scholar] [CrossRef]
- Rupley, J.A.; Careri, G. Protein hydration and function. Adv. Protein Chem. 1991, 41, 37–172. [Google Scholar] [CrossRef] [PubMed]
- Borovykh, V.; Gast, P.; Dzuba, S.A. The dynamical transition in proteins of bacterial photosynthetic reaction centers observed by echo-detected EPR of specific spin labels. Appl. Magn. Reson. 2007, 31, 159–166. [Google Scholar] [CrossRef]
- Möbius, K.; Savitsky, V.; Schnegg, A.; Plato, M.; Fuchs, M. High-field EPR spectroscopy applied to biological systems: Characterization of molecular switches for electron and ion transfer. Phys. Chem. Chem. Phys. 2005, 7, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Leporini, D.; Jeschke, G. Cage effects on the librational motion in ionomeric homopolymers and block copolymers. Phil. Mag. 2004, 84, 1567–1572. [Google Scholar] [CrossRef]
- Brustolon, M.; Barbon, A.; Bortolus, M.; Maniero, A.L.; Sozzani, P.; Comotti, A.; Simonutti, R. Dynamics of Alkoxy-Oligothiophene Ground and Excited States in Nanochannels. J. Am. Chem. Soc. 2004, 126, 15512–15519. [Google Scholar] [CrossRef]
- Dzuba, S.A.; Golovina, Y.A.; Tsvetkov, Y.D. Echo-induced EPR spectra of spin probes as a method for identification of glassy state in biological objects. J. Magn. Reson. B 1993, 101, 134–138. [Google Scholar] [CrossRef]
- Buitink, J.; Dzuba, S.A.; Hoekstra, F.A.; Tsvetkov, Y.D. Pulsed EPR spin-probe study of intracellular glasses in seed and pollen. J. Magn. Reson. 2000, 142, 364–368. [Google Scholar] [CrossRef]
- Böhmer, R.; Diezemann, G.; Hinze, G.; Rössler, E. Dynamics of supercooled liquids and glassy solids. Progr. Nucl. Magn. Reson. Spectr. 2001, 39, 191–267. [Google Scholar] [CrossRef]
- Batchelor, L.S.; Niu, C.N.; Torchia, D.A. Methyl reorientation in polycrystalline amino acids and peptides: A deuteron NMR spin-lattice relaxation study. J. Am. Chem. Soc. 1983, 105, 2228–2231. [Google Scholar] [CrossRef]
- Vogel, M.; Tschirwitz, C.; Schneider, G.; Koplin, C.; Medick, P.; Rössler, E. A 2H NMR and dielelectric spectroscopy study of the slow β-process in organic glass formers. J. Non-Cryst. Solids 2002, 307, 326. [Google Scholar] [CrossRef]
- Pötzschner, B.; Mohamed, F.; Bächer, C.; Wagner, E.; Lichtinger, A.; Bock, D.; Kreger, K.; Schmidt, H.-W.; Rössler, E.A. Non-polymeric asymmetric binary glass-formers. II. Secondary relaxation studied by dielectric, 2H NMR, and 31P NMR spectroscopy. J. Chem. Phys. 2017, 146, 164504. [Google Scholar] [CrossRef]
- Steinrucken, E.; Becher, M.; Vogel, M. On the molecular mechanisms of α and β relaxations in ionic liquids. J. Chem. Phys. 2020, 153, 104507. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Spin-Labeled Molecule | Td | Reference |
|---|---|---|---|
| DPPC/Cholesterol (50:50 mol/mol) membrane | 14-PCSL | 210 K | [120] |
| Human serum albumin | 5-DSA, 16-DSA | 210 K | [123] |
| Alamethicin (Ala) in membrane | Ala/TOAC, with different label position | 160 or 220 K, depending on the position | [124] |
| Human haemoglobin (Hb) in water | Hb/6-MSL | 200 K | [107] |
| Hb in 60% v/v glycerol–water | Hb/6-MSL | 210 K | |
| Hb lyophilized | Hb/6-MSL | non-detectable | |
| Human haemoglobin (Hb) | Hb/6-MSL | 210 K | [125] |
| Human serum albumin (HSA) | HAS/5-MSL | 210 K | |
| β-Lactoglobulin (β-LG) | β-LG/5-MSL | 210 K | |
| β-LG in 60% v/v glycerol–water | β-LG/5-MSL | 240 K | |
| Na,K-ATPase | 5-DSA (14-DSA) | 240 K (200 K) | [126] |
| Na,K-ATPase | Na,K-ATPase/5-MSL | 220 K | |
| DPPC membrane | 5-PCSL (16-PCSL) | 240 K (230 K) | [117] |
| DPPC membrane | 5-PCSL (16-PCSL) | 220 K (210 K) | [127] |
| DHPC membrane (interdigitated chains) | 5-PCSL (16-PCSL) | 240 K (230 K) | |
| POPC membrane | 5-PCSL (16-PCSL) | 240 K (210 K) | [114] |
| DOPC membrane | 5-PCSL (16-PCSL) | 240 K (190 K) | |
| DPPC/Lyso-PPC (interdigitated chains) | 5-PCSL, 16-PCSL | 220 K | [128] |
| DPPC membrane | 14-PCSL | 225 K | [129] |
| DPPC membrane (hydrated) | 4-PCSL | 240 K | |
| DPPC membrane (low hydration) | 4-PCSL | non-detectable | |
| Lysozyme | Lysozyme/IASL | 195 K | [108] |
| Casein | Casein/IASL | 235 K |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dzuba, S.A. Probing Small-Angle Molecular Motions with EPR Spectroscopy: Dynamical Transition and Molecular Packing in Disordered Solids. Magnetochemistry 2022, 8, 19. https://doi.org/10.3390/magnetochemistry8020019
Dzuba SA. Probing Small-Angle Molecular Motions with EPR Spectroscopy: Dynamical Transition and Molecular Packing in Disordered Solids. Magnetochemistry. 2022; 8(2):19. https://doi.org/10.3390/magnetochemistry8020019
Chicago/Turabian StyleDzuba, Sergei A. 2022. "Probing Small-Angle Molecular Motions with EPR Spectroscopy: Dynamical Transition and Molecular Packing in Disordered Solids" Magnetochemistry 8, no. 2: 19. https://doi.org/10.3390/magnetochemistry8020019