Five 2,6-Di(pyrazol-1-yl)pyridine-4-carboxylate Esters, and the Spin States of their Iron(II) Complexes


Abstract
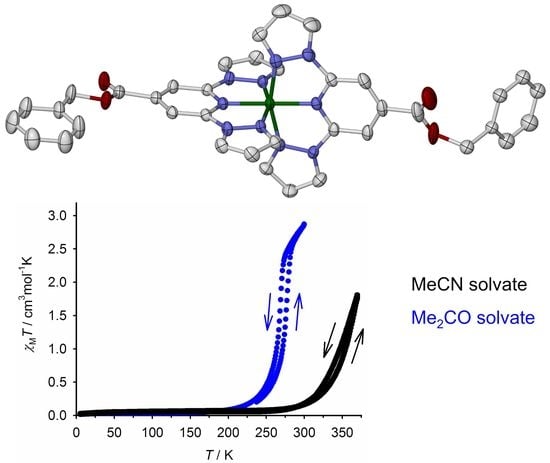
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Crystallographic Characterization
2.3. Characterization of the Bulk Materials
3. Experimental Section
3.1. Instrumentation
3.2. Synthesis
3.2.1. Synthesis of 3,4-Dimethoxyphenyl 2,6-di(pyrazol-1-yl)pyridine-4-carboxylate (L3)
3.2.2. Synthesis of 3,4-Dihydroxyphenyl 2,6-di(pyrazol-1-yl)pyridine-4-carboxylate (L4)
3.2.3. Synthesis of Benzyl 2,6-di(pyrazol-1-yl)pyridine-4-carboxylate (L5)
3.2.4. Synthesis of 4-Methoxybenzyl 2,6-di(pyrazol-1-yl)pyridine-4-carboxylate (L6)
3.2.5. Synthesis of 3,4-Dimethoxybenzyl 2,6-di(pyrazol-1-yl)pyridine-4-carboxylate (L7)
3.2.6. Synthesis of [Fe(L3)2][BF4]2
3.2.7. Synthesis of [Fe(L4)2][BF4]2
3.2.8. Synthesis of [Fe(L5)2][BF4]2
3.2.9. Synthesis of [Fe(L6)2][BF4]2
3.2.10. Synthesis of [Fe(L7)2][BF4]2
3.3. Crystal Structure Determinations
3.3.1. Organic Ligand Crystallographic Refinements
3.3.2. Crystallographic Refinement of α-[Fe(L3)2][BF4]2·2MeNO2
3.3.3. Crystallographic Refinement of β-[Fe(L3)2][BF4]2·2MeNO2
3.3.4. Crystallographic Refinement of [Fe(L4)2][BF4]2
3.3.5. Crystallographic Refinement of [Fe(L5)2][BF4]2·3/2MeCN
3.3.6. Crystallographic Refinement of [Fe(L6)2][BF4]2·3/2MeCN
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gütlich, P.; Goodwin, H.A. (Eds.) Spin Crossover in Transition Metal Compounds I-III. Topics in Current Chemistry Vols. 233–235; Springer: Berlin, Germany, 2004. [Google Scholar]
- Halcrow, M.A. (Ed.) Spin-Crossover Materials—Properties and Applications; John Wiley & Sons: Chichester, UK, 2013; p. 568. [Google Scholar]
- Kumar, K.S.; Ruben, M. Emerging trends in spin crossover (SCO) based functional materials and devices. Coord. Chem. Rev. 2017, 346, 176–205. [Google Scholar] [CrossRef]
- Kahn, O.; Kröber, J.; Jay, C. Spin transition molecular materials for displays and data recording. Adv. Mater. 1992, 4, 718–728. [Google Scholar] [CrossRef]
- Manrique-Juárez, M.D.; Rat, S.; Salmon, L.; Molnár, G.; Quintero, C.M.; Nicu, L.; Shepherd, H.J.; Bousseksou, A. Switchable molecule-based materials for micro- and nanoscale actuating applications: Achievements and prospects. Coord. Chem. Rev. 2016, 308, 395–408. [Google Scholar] [CrossRef]
- Zhang, X.; Mu, S.; Chastanet, G.; Daro, N.; Palamarciuc, T.; Rosa, P.; Létard, J.-F.; Liu, J.; Sterbinsky, G.E.; Arena, D.A.; et al. Complexities in the molecular spin crossover transition. J. Phys. Chem. C 2015, 119, 16293–16302. [Google Scholar] [CrossRef]
- Bovo, G.; Braunlich, I.; Caseri, W.R.; Stingelin, N.; Anthopoulos, T.D.; Sandeman, K.G.; Bradley, D.D.C.; Stavrinou, P.N. Room temperature dielectric bistability in solution-processed spin crossover polymer thin films. J. Mater. Chem. C 2016, 4, 6240–6248. [Google Scholar] [CrossRef]
- Rat, S.; Piedrahita-Bello, M.; Salmon, L.; Molnár, G.; Demont, P.; Bousseksou, A. Coupling mechanical and electrical properties in spin crossover polymer composites. Adv. Mater. 2018, 30, 1705275. [Google Scholar] [CrossRef] [PubMed]
- Diaconu, A.; Lupu, S.-L.; Rusu, I.; Risca, I.-M.; Salmon, L.; Molnár, G.; Bousseksou, A.; Demont, P.; Rotaru, A. Piezoresistive effect in the [Fe(Htrz)2(trz)](BF4) spin crossover complex. J. Phys. Chem. Lett. 2017, 8, 3147–3151. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.; Benjamin, S.M.; Steven, E.; Brooks, J.S.; Shatruk, M. Photomagnetic response in highly conductive iron(II) spin-crossover complexes with TCNQ radicals. Angew. Chem. Int. Ed. 2015, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Shvachko, Y.N.; Starichenko, D.V.; Korolyov, A.V.; Yagubskii, E.B.; Kotov, A.I.; Buravov, L.I.; Lyssenko, K.A.; Zverev, V.N.; Simonov, S.V.; Zorina, L.V.; et al. The conducting spin-crossover compound combining Fe(II) cation complex with TCNQ in a fractional reduction state. Inorg. Chem. 2016, 55, 9121–9130. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Ge, J.-Y.; Hua, C.; Jiao, C.-Q.; Wu, Y.; Leong, C.F.; D’Alessandro, D.M.; Liu, T.; Zuo, J.-L. Photo- and electronically switchable spin-crossover iron(II) metal-organic frameworks based on a tetrathiafulvalene ligand. Angew. Chem. Int. Ed. 2017, 56, 5465–5470. [Google Scholar] [CrossRef]
- Lochenie, C.; Schötz, K.; Panzer, F.; Kurz, H.; Maier, B.; Puchtler, F.; Agarwal, S.; Köhler, A.; Weber, B. Spin-crossover iron(II) coordination polymer with fluorescent properties: Correlation between emission properties and spin state. J. Am. Chem. Soc. 2018, 140, 700–709. [Google Scholar] [CrossRef]
- Yuan, J.; Wu, S.-Q.; Liu, M.-J.; Sato, O.; Kou, H.-Z. Rhodamine 6G-labeled pyridyl aroylhydrazone Fe(II) complex exhibiting synergetic spin crossover and fluorescence. J. Am. Chem. Soc. 2018, 140, 9426–9433. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Li.; Liu, Q.; Meng, Y.-S.; Liu, X.; Zheng, H.; Shi, Q.; Duan, C.-Y.; Liu, T. Fluorescence modulation via photoinduced spin crossover switched energy transfer from fluorophores to FeII ions. Chem. Sci. 2018, 9, 2892–2897. [Google Scholar] [CrossRef]
- Gaspar, A.B.; Seredyuk, M. Spin crossover in soft matter. Coord. Chem. Rev. 2014, 268, 41–58. [Google Scholar] [CrossRef]
- Kuroiwa, K. Supramolecular control of spin crossover phenomena using various amphiphiles. Inorganics 2017, 5, 45. [Google Scholar] [CrossRef]
- Salmon, L.; Catala, L. Spin-crossover nanoparticles and nanocomposite materials. C. R. Chim. 2018, 21, 1230–1269. [Google Scholar] [CrossRef]
- Mallah, T.; Cavallini, M. Surfaces, thin films and patterning of spin crossover compounds. C. R. Chim. 2018, 21, 1270–1286. [Google Scholar] [CrossRef]
- Mikolasek, M.; Felix, G.; Nicolazzi, W.; Molnár, G.; Salmon, L.; Bousseksou, A. Finite size effects in molecular spin crossover materials. New J. Chem. 2014, 38, 1834–1839. [Google Scholar] [CrossRef]
- Molnár, G.; Rat, S.; Salmon, L.; Nicolazzi, W.; Bousseksou, A. Spin crossover nanomaterials: From fundamental concepts to devices. Adv. Mater. 2018, 30, 1703862. [Google Scholar] [CrossRef]
- Halcrow, M.A. Iron(II) complexes of 2,6-di(pyrazol-1-yl)pyridines—A versatile system for spin-crossover research. Coord. Chem. Rev. 2009, 253, 2493–2514. [Google Scholar] [CrossRef]
- Olguín, J.; Brooker, S. Spin crossover active iron(II) complexes of selected pyrazole-pyridine/pyrazine ligands. Coord. Chem. Rev. 2011, 255, 203–240. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Mohammed, R.; Sherborne, G.; Roberts, T.D.; Alvarez, S.; Halcrow, M.A. Spin state behaviour of iron(II)/dipyrazolylpyridine complexes. New insights from crystallographic and solution measurements. Coord. Chem. Rev. 2015, 289–290, 2–12. [Google Scholar] [CrossRef]
- Halcrow, M.A. Recent advances in the synthesis and applications of 2,6-dipyrazolylpyridine derivatives and their complexes. New J. Chem. 2014, 38, 1868–1882. [Google Scholar]
- Holland, J.M.; Barrett, S.A.; Kilner, C.A.; Halcrow, M.A. Control of the spin state of Fe(II) 2,6-di(pyrazol-1-yl)pyridine complexes by distal ligand substitution. Inorg. Chem. Commun. 2002, 5, 328–332. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Kulmaczewski, R.; Mohammed, R.; Dudley, S.; Barrett, S.A.; Little, M.A.; Deeth, R.J.; Halcrow, M.A. A unified treatment of the relationship between ligand substituents and spin state in a family of iron(II) complexes. Angew. Chem. Int. Ed. 2016, 55, 4327–4331. [Google Scholar] [CrossRef]
- González-Prieto, R.; Fleury, B.; Schramm, F.; Zoppellaro, G.; Chandrasekar, R.; Fuhr, O.; Lebedkin, S.; Kappes, M.; Ruben, M. Tuning the spin-transition properties of pyrene-decorated 2,6-bispyrazolylpyridine based Fe(II) complexes. Dalton Trans. 2011, 40, 7564–7570. [Google Scholar] [CrossRef]
- Kumar, K.S.; Šalitroš, I.; Moreno-Pineda, E.; Ruben, M. Spacer type mediated tunable spin crossover (SCO) characteristics of pyrene decorated 2,6-bis(pyrazol-1-yl)pyridine (bpp) based Fe(II) molecular spintronic modules. Dalton Trans. 2017, 46, 9765–9768. [Google Scholar] [CrossRef]
- Santoro, A.; Kershaw Cook, L.J.; Kulmaczewski, R.; Barrett, S.A.; Cespedes, O.; Halcrow, M.A. Iron(II) complexes of tridentate indazolylpyridine ligands: Enhanced spin-crossover hysteresis and ligand-based fluorescence. Inorg. Chem. 2015, 54, 682–693. [Google Scholar] [CrossRef]
- Schäfer, B.; Bauer, T.; Faus, I.; Wolny, J.A.; Dahms, F.; Fuhr, O.; Lebedkin, S.; Wille, H.-C.; Schlage, K.; Chevalier, K.; et al. A luminescent Pt2Fe spin crossover complex. Dalton Trans. 2017, 46, 2289–2302. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Sakamoto, R.; Takahashi, K.; Nishihara, H. Solid-state ligand-driven light-induced spin change at ambient temperatures in bis(dipyrazolylstyrylpyridine)iron(II) complexes. Inorg. Chem. 2013, 52, 1658–1665. [Google Scholar] [CrossRef]
- Nihei, M.; Han, L.; Oshio, H. Magnetic bistability and single-crystal-to-single-crystal transformation induced by guest desorption. J. Am. Chem. Soc. 2007, 129, 5312–5313. [Google Scholar] [CrossRef] [PubMed]
- Nihei, M.; Takahashi, N.; Nishikawa, H.; Oshio, H. Spin-crossover behavior and electrical conduction property on iron(II) complexes with tetrathiafulvalene moieties. Dalton Trans. 2011, 40, 2154–2156. [Google Scholar] [CrossRef]
- Abhervé, A.; Palacios-Corella, M.; Clemente-Juan, J.M.; Marx, R.; Neugebauer, P.; van Slageren, J.; Clemente-León, M.; Coronado, E. Bimetallic MnIII–FeII hybrid complexes formed by a functionalized MnIII Anderson polyoxometalate coordinated to FeII: Observation of a field-induced slow relaxation of magnetization in the MnIII centres and a photoinduced spin-crossover in the FeII centres. J. Mater. Chem. C 2015, 3, 7936–7945. [Google Scholar] [CrossRef]
- Abhervé, A.; Recio-Carretero, M.J.; López-Jordà, M.; Clemente-Juan, J.M.; Canet-Ferrer, J.; Cantarero, A.; Clemente-León, M.; Coronado, E. Nonanuclear spin-crossover complex containing iron(II) and iron(III) based on a 2,6-bis(pyrazol-1-yl)pyridine ligand functionalized with a carboxylate group. Inorg. Chem. 2016, 55, 9361–9367. [Google Scholar] [CrossRef]
- Rajadurai, C.; Fuhr, O.; Kruk, R.; Ghafari, M.; Hahn, H.; Ruben, M. Above room temperature spin transition in a metallo-supramolecular coordination oligomer/polymer. Chem. Commun. 2007, 2636–2638. [Google Scholar] [CrossRef] [PubMed]
- Tovee, C.A.; Kilner, C.A.; Barrett, S.A.; Thomas, J.A.; Halcrow, M.A. A back-to-back ligand with dipyrazolylpyridine and dipicolylamine metal-binding domains. Eur. J. Inorg. Chem. 2010, 2010, 1007–1012. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Fisher, J.; Harding, L.P.; Halcrow, M.A. An iron(II) spin-crossover metallacycle from a back-to-back bis-[dipyrazolylpyridine]. Dalton Trans. 2015, 44, 9417–9425. [Google Scholar] [CrossRef]
- Devid, E.J.; Martinho, P.N.; Kamalakar, M.V.; Šalitroš, I.; Prendergast, U.; Dayen, J.-F.; Meded, V.; Lemma, T.; González-Prieto, R.; Evers, F.; et al. Spin transition in arrays of gold nanoparticles and spin crossover molecules. ACS Nano 2015, 9, 4496–4507. [Google Scholar] [CrossRef]
- Pukenas, L.; Benn, F.; Lovell, E.; Santoro, A.; Kershaw Cook, L.J.; Halcrow, M.A.; Evans, S.D. Bead-like structures and self-assembled monolayers from 2,6-dipyrazolylpyridines and their iron(II) complexes. J. Mater. Chem. C 2015, 3, 7890–7896. [Google Scholar] [CrossRef]
- Kumar, K.S.; Šalitroš, I.; Boubegtiten-Fezoua, Z.; Moldovan, S.; Hellwig, P.; Ruben, M. A spin crossover (SCO) active graphene-iron(II) complex hybrid material. Dalton Trans. 2018, 47, 35–40. [Google Scholar] [CrossRef]
- Galadzhun, I.; Kulmaczewski, R.; Cespedes, O.; Yamada, M.; Yoshinari, N.; Konno, T.; Halcrow, M.A. 2,6-Di(pyrazolyl)pyridine-4-carboxylate esters with alkyl chain substituents, and their iron(II) complexes. Inorg. Chem. 2018, 57, 13761–13771. [Google Scholar] [CrossRef] [PubMed]
- Bridonneau, N.; Rigamonti, L.; Poneti, G.; Pinkowicz, D.; Forni, A.; Cornia, A. Evidence of crystal packing effects in stabilizing high or low spin states of iron(II) complexes with functionalized 2,6-bis(pyrazol-1-yl)pyridine ligands. Dalton Trans. 2017, 46, 4075–4085. [Google Scholar] [CrossRef] [PubMed]
- García-López, V.; Palacios-Corella, M.; Abhervé, A.; Pellicer-Carreño, I.; Desplanches, C.; Clemente-León, M.; Coronado, E. Spin-crossover compounds based on iron(II) complexes of 2,6-bis(pyrazol-1-yl)pyridine (bpp) functionalized with carboxylic acid and ethyl carboxylic acid. Dalton Trans. 2018, 47, 16958–16968. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.S.; Šalitroš, I.; Suryadevara, N.; Moreno-Pineda, E.; Ruben, M. Supramolecular interaction tuning of spin-crossover in pyrene/fullerene (C60) tethered FeII-2,6-di(pyrazol-1-yl)pyridine complexes: Towards switchable molecular devices. Eur. J. Inorg. Chem. 2018, 2018, 5091–5097. [Google Scholar] [CrossRef]
- Vermonden, T.; Branowska, D.; Marcelis, A.T.M.; Sudhölter, E.J.R. Synthesis of 4-functionalized terdendate pyridine-based ligands. Tetrahedron 2003, 59, 5039–5045. [Google Scholar] [CrossRef]
- Halcrow, M.A. The synthesis and coordination chemistry of 2,6-bis(pyrazolyl)pyridines and related ligands—Versatile terpyridine analogues. Coord. Chem. Rev. 2005, 249, 2880–2908. [Google Scholar] [CrossRef]
- Bessel, C.A.; See, R.F.; Jameson, D.L.; Churchill, M.R.; Takeuchi, K.J. Structural considerations of terdentate ligands: Crystal structures of 2,2′:6′,2″-terpyridine and 2,6-bis(pyrazol-1-yl)pyridine. J. Chem. Soc. Dalton Trans. 1992, 3223–3228. [Google Scholar] [CrossRef]
- Guionneau, P.; Marchivie, M.; Bravic, G.; Létard, J.-F.; Chasseau, D. Structural aspects of spin crossover. example of the [FeIILn(NCS)2] complexes. Top. Curr. Chem. 2004, 234, 97–128. [Google Scholar]
- Holland, J.M.; McAllister, J.A.; Kilner, C.A.; Thornton-Pett, M.; Bridgeman, A.J.; Halcrow, M.A. Stereochemical effects on the spin state transition shown by salts of [FeL2]2+ [L = 2,6-di(pyrazol-1-yl)pyridine]. J. Chem. Soc. Dalton Trans. 2002, 548–554. [Google Scholar] [CrossRef]
- Vela, S.; Novoa, J.J.; Ribas-Arino, J. Insights into the crystal-packing effects on the spin crossover of [FeII(1-bpp)2]2+-based materials. Phys. Chem. Chem. Phys. 2014, 16, 27012–27024. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Thorp-Greenwood, F.L.; Comyn, T.P.; Cespedes, O.; Chastanet, G.; Halcrow, M.A. Unexpected spin-crossover and a low pressure phase change in an iron(II)/dipyrazolylpyridine complex exhibiting a high-spin Jahn-Teller distortion. Inorg. Chem. 2015, 54, 6319–6330. [Google Scholar] [CrossRef]
- McCusker, J.K.; Rheingold, A.L.; Hendrickson, D.N. Variable-temperature studies of laser-initiated 5T2 → 1A1 intersystem crossing in spin-crossover complexes: Empirical correlations between activation parameters and ligand structure in a series of polypyridyl ferrous complexes. Inorg. Chem. 1996, 35, 2100–2112. [Google Scholar] [CrossRef]
- Capel Berdiell, I.; Kulmaczewsk, R.; Halcrow, M.A. Iron(II) complexes of 2,4-dipyrazolyl-1,3,5-triazine derivatives—The influence of ligand geometry on metal ion spin state. Inorg. Chem. 2017, 56, 8817–8828. [Google Scholar] [CrossRef]
- O’Connor, C.J. Magnetochemistry—Theory and experimentation. Prog. Inorg. Chem. 1982, 29, 203–283. [Google Scholar]
- Evans, D.F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Schubert, E.M. Utilizing the Evans method with a superconducting NMR spectrometer in the undergraduate laboratory. J. Chem. Educ. 1992, 69, 62. [Google Scholar] [CrossRef]
- García, B.; Ortega, J.C. Excess viscosity ηE, excess volume VE and excess free energy of activation ΔG*E at 283, 293, 303, 313, and 323 K for mixtures of acetonitrile and alkyl benzoates. J. Chem. Eng. Data 1988, 33, 200–204. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Barbour, L.J. X-Seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Tao, J.; Wei, R.-J.; Huang, R.-B.; Zheng, L.-S. Polymorphism in spin-crossover systems. Chem. Soc. Rev. 2012, 41, 703–737. [Google Scholar] [CrossRef]
- Haryono, M.; Heinemann, F.W.; Petukhov, K.; Gieb, K.; Müller, P.; Grohmann, A. Parallel crystallization of a “static” and a spin-crossover polymorph of an iron(II) complex from the same solution. Eur. J. Inorg. Chem. 2009, 2136–2143. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α-[Fe(L3)2][BF4]2·2MeNO2 | β-[Fe(L3)2][BF4]2·2MeNO2 | |
|---|---|---|
| Fe–N{pyridyl} | 1.8903(17), 1.8917(17) | 2.143(3), 2.156(4) |
| Fe–N{pyrazolyl} | 1.9502(19)–1.9831(19) | 2.151(3)–2.208(4) |
| VOh | 9.446(5) | 12.357(13) |
| Σ | 84.6(3) | 159.2(4) |
| Θ | 275 | 475 |
| φ | 174.29(8) | 166.41(12) |
| θ | 89.79(2) | 79.90(3) |
| Compound | [Fe(L4)2][BF4]2 | [Fe(L5)2][BF4]2·3/2MeCN | ||
|---|---|---|---|---|
| Molecule | Molecule A | Molecule B | Molecule A | Molecule B |
| Fe–N{pyridyl} | 1.897(3), 1.897(3) | 1.888(3), 1.898(3) | 1.892(2), 1.892(2) | 1.890(2), 1.891(2) |
| Fe–N{pyrazolyl} | 1.956(4)–1.990(4) | 1.965(4)–1.977(4) | 1.955(2)–1.964(3) | 1.953(3)–1.972(3) |
| VOh | 9.537(11) | 9.499(11) | 9.392(7) | 9.405(8) |
| VOh | 88.8(5) | 84.8(5) | 84.3(4) | 83.6(4) |
| Σ | 275 | 278 | 276 | 274 |
| Θ | 176.93(16) | 178.09(16) | 175.96(11) | 178.45(11) |
| φ | 85.54(5) | 87.63(4) | 88.14(3) | 87.78(3) |
| Compound | [Fe(L6)2][BF4]2·3/2MeCN | |||
| Molecule | Molecule A | Molecule B | ||
| Fe–N{pyridyl} | 1.889(3), 1.896(3) | 1.891(3), 1.897(3) | ||
| Fe–N{pyrazolyl} | 1.965(4)–1.978(3) | 1.963(4)–1.996(4) | ||
| VOh | 9.501(10) | 9.538(11) | ||
| Σ | 83.7(5) | 90.5(5) | ||
| Θ | 277 | 274 | ||
| φ | 176.34(15) | 175.93(15) | ||
| θ | 89.77(3) | 83.18(4) | ||
| Compound | L3 | L5 | L6 | L7·½MeCN |
|---|---|---|---|---|
| formula | C20H17N5O4 | C19H15N5O2 | C20H17N5O3 | C22H20.5N5.5O4 |
| Mr | 391.39 | 345.36 | 375.39 | 425.94 |
| crystal class | monoclinic | monoclinic | orthorhombic | monoclinic |
| space group | P21/c | Pc | Pbca | P21/c |
| a/Å | 11.9677(2) | 7.2833(1) | 42.4116(3) | 8.0803(1) |
| b/Å | 20.7834(4) | 7.7290(1) | 11.4438(1) | 65.5346(6) |
| c/Å | 7.3138(1) | 29.9811(5) | 7.2071(1) | 7.6027(1) |
| β/° | 90.900(2) | 92.924(1) | ‒ | 91.061(1) |
| V/Å3 | 1818.93(5) | 1685.52(4) | 3497.96(6) | 4025.24(8) |
| Z | 4 | 4 | 8 | 8 |
| T/K | 120(2) | 100(2) | 100(2) | 100(2) |
| Dcalcd/Mgm−3 | 1.429 | 1.361 | 1.426 | 1.406 |
| μ/mm−1 | 0.855 [a] | 0.093 [b] | 0.100 [b] | 0.100 [b] |
| measured reflections | 7444 | 25,320 | 50,109 | 62,089 |
| unique reflections | 3524 | 7820 | 8534 | 18,798 |
| observed reflections | 3020 | 4902 | 6291 | 12,124 |
| Rint | 0.026 | 0.102 | 0.091 | 0.056 |
| R1 [Fo > 4σ(Fo)] [c] | 0.042 | 0.070 | 0.054 | 0.063 |
| wR2 [all data] [d] | 0.113 | 0.192 | 0.162 | 0.174 |
| GoF | 1.038 | 0.992 | 1.021 | 1.034 |
| Flack parameter | ‒ | ‒0.5(12) [e] | ‒ | ‒ |
| Δρmax, Δρmin/eÅ−3 | 0.31/‒0.21 | 0.34/‒0.44 | 0.80/‒0.35 | 0.56/‒0.42 |
| Compound | α-[Fe(L3)2][BF4]2-2MeNO2 | β-[Fe(L3)2][BF4]2-2MeNO2 | [Fe(L4)2][BF4]2 |
|---|---|---|---|
| formula | C42H40B2F8FeN12O12 | C42H40B2F8FeN12O12 | C36H26B2F8FeN10O8 |
| Mr | 1134.33 | 1134.33 | 956.14 |
| crystal class | triclinic | triclinic | triclinic |
| space group | P | P | P |
| a/Å | 8.3230(2) | 11.3601(9) | 8.6675(3) |
| b/Å | 12.8302(2) | 13.2652(13) | 16.8378(6) |
| c/Å | 23.1765(6) | 17.5490(15) | 27.0770(11) |
| α/° | 88.116(2) | 67.671(9) | 85.102(3) |
| β/° | 80.270(2) | 80.155(7) | 82.500(3) |
| γ/° | 89.567(2) | 78.463(8) | 87.451(3) |
| V/Å3 | 2438.00(9) | 2383.6(4) | 3901.3(3) |
| Z | 2 | 2 | 4 |
| T/K | 120(2) | 120(2) | 120(2) |
| Dcalcd/Mgm−3 | 1.545 | 1.580 | 1.628 |
| μ/mm−1 | 3.412 [a] | 3.490 [a] | 4.048 [a] |
| measured reflections | 42,688 | 18,126 | 59,552 |
| unique reflections | 9491 | 8979 | 15,161 |
| observed reflections | 9123 | 6467 | 10,540 |
| Rint | 0.035 | 0.065 | 0.075 |
| R1 [Fo > 4 σ(Fo)] [c] | 0.048 | 0.068 | 0.073 |
| wR2 [all data] [d] | 0.134 | 0.186 | 0.218 |
| GoF | 1.066 | 1.049 | 1.030 |
| Δρmax, Δρmin/eÅ−3 | 1.30/‒0.85 | 0.71/‒0.84 | 0.85/‒0.61 |
| Compound | [Fe(L5)2][BF4]2·3/2MeCN | [Fe(L6)2][BF4]2·3/2MeCN | |
| formula | C41H34.5B2F8FeN11.5O4 | C43H38.5B2F8FeN11.5O6 | |
| Mr | 981.77 | 1041.82 | |
| crystal class | triclinic | monoclinic | |
| space group | P | P21/c | |
| a/Å | 13.8965(6) | 27.4401(4) | |
| b/Å | 17.0836(7) | 16.5615(3) | |
| c/Å | 20.8001(7) | 20.5281(2) | |
| α/° | 95.020(3) | ‒ | |
| β/° | 102.703(3) | 100.257(1) | |
| γ/° | 113.133(4) | ‒ | |
| V/Å3 | 4345.8(3) | 9179.9(2) | |
| Z | 4 | 8 | |
| T/K | 150(2) | 150(2) | |
| Dcalcd/Mgm−3 | 1.501 | 1.508 | |
| μ/mm−1 | 0.438 [b] | 3.466 [a] | |
| measured reflections | 46,531 | 77,065 | |
| unique reflections | 20,799 | 17,911 | |
| observed reflections | 13,895 | 15,370 | |
| Rint | 0.037 | 0.051 | |
| R1 [Fo > 4σ(Fo)] [c] | 0.067 | 0.080 | |
| wR2 [all data] [d] | 0.170 | 0.220 | |
| GoF | 1.032 | 1.033 | |
| Δρmax, Δρmin/eÅ−3 | 1.02/‒0.59 | 1.25/‒0.97 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galadzhun, I.; Kulmaczewski, R.; Halcrow, M.A. Five 2,6-Di(pyrazol-1-yl)pyridine-4-carboxylate Esters, and the Spin States of their Iron(II) Complexes. Magnetochemistry 2019, 5, 9. https://doi.org/10.3390/magnetochemistry5010009
Galadzhun I, Kulmaczewski R, Halcrow MA. Five 2,6-Di(pyrazol-1-yl)pyridine-4-carboxylate Esters, and the Spin States of their Iron(II) Complexes. Magnetochemistry. 2019; 5(1):9. https://doi.org/10.3390/magnetochemistry5010009
Chicago/Turabian StyleGaladzhun, Iurii, Rafal Kulmaczewski, and Malcolm A. Halcrow. 2019. "Five 2,6-Di(pyrazol-1-yl)pyridine-4-carboxylate Esters, and the Spin States of their Iron(II) Complexes" Magnetochemistry 5, no. 1: 9. https://doi.org/10.3390/magnetochemistry5010009