
Transcriptome Analysis of Berries of Spine Grape (Vitis davidii Föex) Infected by Colletotrichum viniferum during Symptom Development


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. RNA Extraction and Transcriptome Analysis
2.3. Functional Annotation of Differentially Expressed Genes (DEGs)
2.4. Co-Expression Analysis
2.5. Quantitative RT-PCR (qRT-PCR) Analysis
3. Results
3.1. Statistics of Infected Berries Inoculated with Colletotrichum Viniferum
3.2. Transcriptome Sequencing and Data Statistics
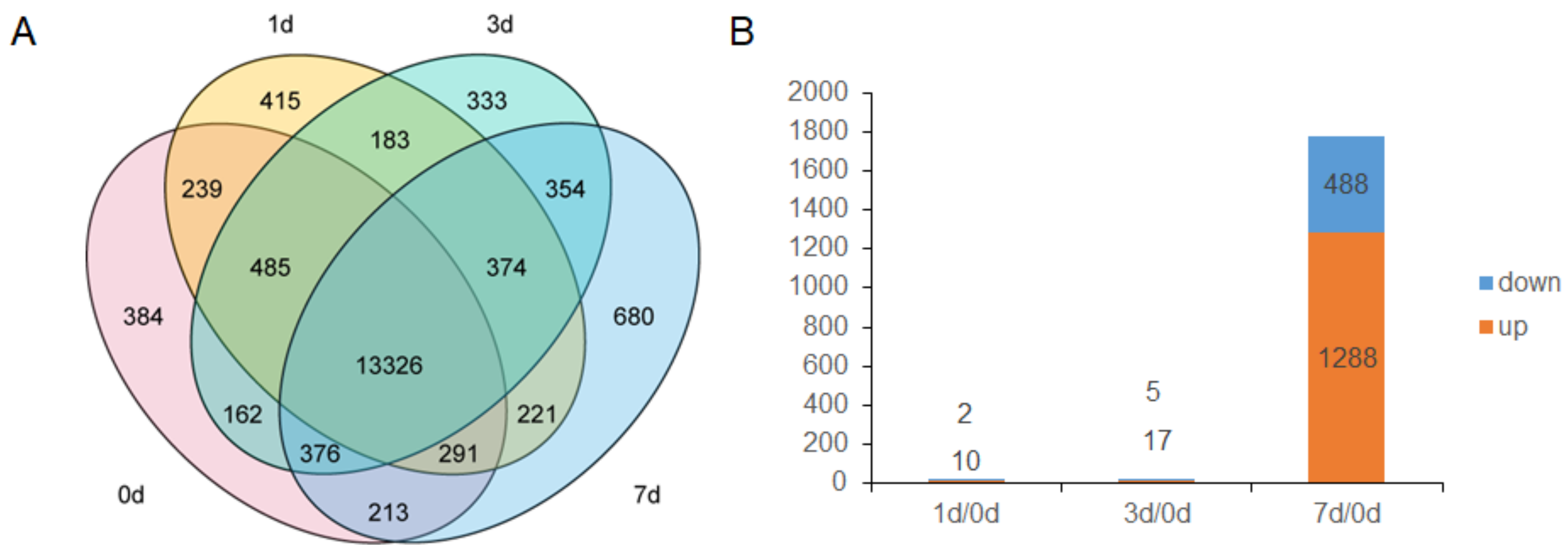
3.3. DEGs of V. davidii after Infection
3.4. GO Functional Analysis of the DEGs
3.5. KEGG Pathway Enrichment Analysis of the DEGs
3.6. DEGs Involved in Primary Metabolism
3.7. DEGs Involved in Signal Transduction
3.8. DEGs Involved in Pathogen-Resistance Genes
3.9. DEGs Involved in Phenylalanine Metabolism Pathway
3.10. Co-Expression Analysis of Differentially Expressed Transcription Factors and Target Functional Genes
3.11. Validation of Differentially Expressed Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Welke, J.E. Fungal and mycotoxin problems in grape juice and wine industries. Curr. Opin. Food Sci. 2019, 29, 7–13. [Google Scholar] [CrossRef]
- Oo, M.M.; Oh, S. Identification and characterization of new record of grape ripe rot disease caused by Colletotrichum viniferum in Korea. Mycobiology 2017, 45, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Cosseboom, S.D.; Hu, M. Predicting Ripe Rot of Grape, Caused by Colletotrichum fioriniae, with Leaf Wetness, Temperature, and the Crop Growth. PhytoFrontiers 2022. [Google Scholar] [CrossRef]
- Whitelawweckert, M.A.; Curtin, S.J.; Huang, R.; Steel, C.; Blanchard, C.; Roffey, P. Phylogenetic relationships and pathogenicity of Colletotrichum acutatum isolates from grape in subtropical Australia. Plant Pathol. 2007, 56, 448–463. [Google Scholar] [CrossRef]
- Dou, M.; Hao, Y.; Yang, J.; Yuan, X.; Yin, X.; Jiao, Y.; Zhao, J.; Chen, T.; Wang, Y.; Xu, Y. Genome Sequence Resource for Colletotrichum viniferum, the cause of grapevine ripe rot in China. Mol. Plant-Microbe Interact. 2022, 35, 90–93. [Google Scholar] [CrossRef]
- Sadoughi, N. Effect of Ripe Rot of Grapes (Colletotrichum spp.) on the Chemical Composition and Off-Flavour Compounds in Grapes and Wine. Ph.D. Dissertation, Charles Sturt University, Bathurst, Australia, 2016. [Google Scholar]
- Ji, T.; Salotti, I.; Dong, C.; Li, M.; Rossi, V. Modeling the effects of the environment and the host plant on the ripe rot of grapes, caused by the Colletotrichum species. Plants 2021, 10, 2288. [Google Scholar] [CrossRef]
- Shiraishi, M.; Koide, M.; Itamura, H.; Yamada, M.; Mitani, N.; Ueno, T.; Nakaune, R.; Nakano, M. Screening for resistance to ripe rot caused by Colletotrichum acutatum in grape germplasm. Vitis 2007, 46, 196–201. [Google Scholar]
- Fu, P.; Tian, Q.; Lai, G.; Li, R.; Song, S.; Lu, J. Cgr1, a ripe rot resistance QTL in Vitis amurensis ‘Shuang Hong’ grapevine. Hortic. Res. 2019, 6, 67. [Google Scholar] [CrossRef]
- Reid, K.E.; Olsson, N.; Schlosser, J.; Peng, F.Y.; Lund, S.T. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 2006, 6, 27. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B.J. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef]
- Kotera, M.; Hirakawa, M.; Tokimatsu, T.; Goto, S.; Kanehisa, M. The KEGG databases and tools facilitating omics analysis: Latest developments involving human diseases and pharmaceuticals. Methods Mol. Biol. 2012, 802, 19–39. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sudheeran, P.K.; Sela, N.; Carmeli-Weissberg, M.; Ovadia, R.; Panda, S.; Feygenberg, O.; Maurer, D.; Oren-Shamir, M.; Aharoni, A.; Alkan, N. Induced defense response in red mango fruit against Colletotrichum gloeosporioides. Hortic. Res. 2021, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, J.; Yin, L.; Zhang, Y.; Qu, J.; Lu, J. Comparative transcriptome analysis reveals defense-related genes and pathways against downy mildew in Vitis amurensis grapevine. Plant Physiol. Biochem. 2015, 95, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, R.; Zhang, N.; Ma, F.; Jiao, Y.; Wang, Z. Transcriptome profiling of Vitis amurensis, an extremely cold-tolerant Chinese wild Vitis species, reveals candidate genes and events that potentially connected to cold stress. Plant Mol. Biol. 2014, 86, 527–541. [Google Scholar] [CrossRef]
- Marzin, S.; Hanemann, A.; Sharma, S.; Hensel, G.; Kumlehn, J.; Schweizer, G.; Roder, M.S. Are PECTIN ESTERASE INHIBITOR genes involved in mediating resistance to rhynchosporium commune in barley. PLoS ONE 2016, 11, e0150485. [Google Scholar] [CrossRef]
- Baker, B.; Zambryski, P.; Staskawicz, B.J.; Dineshkumar, S.P. Signaling in plant-microbe interactions. Science 1997, 276, 726–733. [Google Scholar] [CrossRef]
- Freeman, S.; Katan, T.; Shabi, E. Characterization of Colletotrichum species responsible for anthracnose diseases of various fruits. Plant Dis. 1998, 82, 596–605. [Google Scholar] [CrossRef]
- Jayawardena, R.S.; Hyde, K.D.; Damm, U.; Cai, L.; Liu, M.; Li, X.; Zhang, W.; Zhao, W.; Yan, J.Y. Notes on currently accepted species of Colletotrichum. Mycosphere 2016, 7, 1192–1260. [Google Scholar] [CrossRef]
- Hong, S.; Kim, W.; Yun, H.; Choi, K. Morphological variations, genetic diversity and pathogenicity of Colletotrichum species causing grape ripe rot in Korea. Plant Pathol. J. 2008, 24, 269–278. [Google Scholar] [CrossRef]
- Echeverrigaray, S.; Scariot, F.J.; Fontanella, G.; Favaron, F.; Sella, L.; Santos, M.C.; Schwambach, J.; Pedrotti, C.; Delamare, A.P. Colletotrichum species causing grape ripe rot disease in Vitis labrusca and V. vinifera varieties in the highlands of southern Brazil. Plant Pathol. 2020, 69, 1504–1512. [Google Scholar] [CrossRef]
- Yan, J.; Jayawardena, M.M.R.S.; Goonasekara, I.D.; Wang, Y.; Zhang, W.; Liu, M.; Huang, J.; Wangm, Z.; Shang, J.; Peng, Y. Diverse species of Colletotrichum associated with grapevine anthracnose in China. Fungal Divers. 2014, 71, 233–246. [Google Scholar] [CrossRef]
- Kim, J.S.; Hassan, O.; Chang, T. First report of Colletotrichum aenigma causing anthracnose of grape in Korea. Plant Dis. 2021, 105, 2729. [Google Scholar] [CrossRef] [PubMed]
- Sawant, I.S.; Wadkar, P.N.; Rajguru, Y.R.; Mhaske, N.H.; Salunkhe, V.P.; Sawant, S.D.; Upadhyay, A. Biocontrol potential of two novel grapevine associated Bacillus strains for management of anthracnose disease caused by Colletotrichum gloeosporioides. Biocontrol. Sci. Technol. 2016, 26, 964–979. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, X.B.; Jayawardena, R.S.; Yan, J.Y.; Wang, X.D.; Liu, M.; Chen, T.; Liu, X.M.; Wang, J.C.; Chen, Q.X. Identification and characterization of Colletotrichum species causing grape ripe rot in southern China. Mycosphere 2016, 7, 1177–1191. [Google Scholar] [CrossRef]
- Fung, R.W.M.; Gonzalo, M.; Fekete, C.; Kovacs, L.; He, Y.; Marsh, E.L.; Mcintyre, L.M.; Schachtman, D.P.; Qiu, W. Powdery mildew induces defense-oriented reprogramming of the transcriptome in a susceptible but not in a resistant grapevine. Plant Physiol. 2007, 146, 236–249. [Google Scholar] [CrossRef]
- Bhadauria, V.; Bett, K.E.; Zhou, T.; Vandenberg, A.; Wei, Y.; Banniza, S. Identification of Lens culinaris defense genes responsive to the anthracnose pathogen Colletotrichum truncatum. BMC Genet. 2013, 14, 31. [Google Scholar] [CrossRef]
- Padder, B.A.; Kamfwa, K.; Awale, H.; Kelly, J.D. Transcriptome profiling of the phaseolus vulgaris-Colletotrichum lindemuthianum pathosystem. PLoS ONE 2016, 11, e0165823. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Cao, H.; Hao, X.; Zeng, J.; Yang, Y.; Wang, X. Transcriptome analysis of an anthracnose-resistant tea plant cultivar reveals genes associated with resistance to Colletotrichum camelliae. PLoS ONE 2016, 11, e0148535. [Google Scholar] [CrossRef]
- Mishra, R.; Nanda, S.; Rout, E.; Chand, S.K.; Mohanty, J.N.; Joshi, R.K. Differential expression of defense-related genes in chilli pepper infected with anthracnose pathogen Colletotrichum truncatum. Physiol. Mol. Plant Pathol. 2017, 97, 1–10. [Google Scholar] [CrossRef]
- Wang, Y.; Hao, X.; Lu, Q.; Wang, L.; Qian, W.; Li, N.; Ding, C.; Wang, X.; Yang, Y. Transcriptional analysis and histochemistry reveal that hypersensitive cell death and H2O2 have crucial roles in the resistance of tea plant (Camellia sinensis (L.) O. Kuntze) to anthracnose. Hortic. Res. 2018, 5, 18. [Google Scholar] [CrossRef]
- Trouvelot, S.; Heloir, M.; Poinssot, B.; Gauthier, A.; Paris, F.; Guillier, C.; Combier, M.; Trda, L.; Daire, X.; Adrian, M. Carbohydrates in plant immunity and plant protection: Roles and potential application as foliar sprays. Front. Plant Sci. 2014, 5, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Wang, W.; Xie, Q.; Liu, N.; Liu, L.; Wang, D.; Zhang, X.; Yang, C.; Chen, X.; Tang, D. Plants transfer lipids to sustain colonization by mutualistic mycorrhizal and parasitic fungi. Science 2017, 356, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Cervone, F.; Bellincampi, D. Methyl esterification of pectin plays a role during plant–pathogen interactions and affects plant resistance to diseases. J. Plant Physiol. 2012, 169, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Li, W.; Mi, B.; Ma, Z.; Dawuda, M.M.; Zuo, C.; Zhang, Y.; Jiang, X.; Chen, B. Transcriptome analysis revealed glucose application affects plant hormone signal transduction pathway in “Red Globe” grape plantlets. Plant Growth Regul. 2018, 84, 45–56. [Google Scholar] [CrossRef]
- Dixon, R.A.; Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant Cell 1995, 7, 1085–1097. [Google Scholar] [CrossRef]
- Patel, R.R.; Patel, D.D.; Bhatt, J.; Thakor, P.; Triplett, L.R.; Thakkar, V.R. Induction of pre-chorismate, jasmonate and salicylate pathways by Burkholderia sp. RR18 in peanut seedlings. J. Appl. Microbiol. 2021, 131, 1417–1430. [Google Scholar] [CrossRef]
- Dixon, R.A. Natural products and plant disease resistance. Nature 2001, 411, 843–847. [Google Scholar] [CrossRef]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef]
- Ciaffi, M.; Paolacci, A.R.; Paolocci, M.; Alicandri, E.; Bigini, V.; Badiani, M.; Muganu, M. Transcriptional regulation of stilbene synthases in grapevine germplasm differentially susceptible to downy mildew. BMC Plant Biol. 2019, 19, 404. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, S. MAPK cascades in plant disease resistance signaling. Annu. Rev. Phytopathol. 2013, 51, 245–266. [Google Scholar] [CrossRef]
- Guo, W.W.; Jin, L.; Miao, Y.; He, X.; Hu, Q.; Guo, K.; Zhu, L.; Zhang, X. An ethylene response-related factor, GbERF1-like, from Gossypium barbadense improves resistance to Verticillium dahliae via activating lignin synthesis. Plant Mol. Biol. 2016, 91, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.C.J.; Matus, J.T. Constructing integrated networks for identifying new secondary metabolic pathway regulators in grapevine: Recent applications and future opportunities. Front. Plant Sci. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vannozzi, A.; Wong, D.C.J.; Holl, J.; Hmmam, I.; Matus, J.T.; Bogs, J.; Ziegler, T.; Dry, I.B.; Barcaccia, G.; Lucchin, M. Combinatorial regulation of stilbene synthase genes by WRKY and MYB transcription factors in grapevine (Vitis vinifera L.). Plant Cell Physiol. 2018, 59, 1043–1059. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Data (bp) | Q30 (%) | All Reads Num | Unique Mapped Reads | Mapping Ratio |
|---|---|---|---|---|---|
| CK-1 | 4,018,242,900 | 94.71% | 26,705,700 | 21,281,489 | 86.92% |
| CK-2 | 3,243,124,800 | 92.76% | 21,536,268 | 16,703,124 | 84.22% |
| CK-3 | 3,717,704,100 | 92.20% | 24,716,638 | 19,629,622 | 85.51% |
| 1 dpi-1 | 4,110,842,400 | 92.49% | 27,270,040 | 21,660,483 | 86.46% |
| 1 dpi-2 | 3,827,514,000 | 92.69% | 25,395,464 | 19,739,443 | 84.27% |
| 1 dpi-3 | 3,105,880,500 | 92.77% | 20,600,182 | 15,925,781 | 83.94% |
| 3 dpi-1 | 3,842,454,600 | 94.52% | 25,584,844 | 20,578,578 | 87.13% |
| 3 dpi-2 | 3,693,470,400 | 92.63% | 24,576,840 | 19,248,684 | 85.01% |
| 3 dpi-3 | 4,030,943,700 | 92.71% | 26,802,666 | 21,077,493 | 85.21% |
| 7 dpi-1 | 3,517,864,800 | 92.85% | 23,405,352 | 17,628,147 | 82.03% |
| 7 dpi-2 | 3,743,716,200 | 92.99% | 24,914,150 | 18,468,163 | 80.75% |
| 7 dpi-3 | 4,558,513,200 | 94.94% | 30,292,856 | 21,269,288 | 75.81% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, Y.; Yuan, X.; Chen, T.; Yuan, Y.; Liu, X.; Tang, X.; Chen, Q. Transcriptome Analysis of Berries of Spine Grape (Vitis davidii Föex) Infected by Colletotrichum viniferum during Symptom Development. Horticulturae 2022, 8, 843. https://doi.org/10.3390/horticulturae8090843
Lei Y, Yuan X, Chen T, Yuan Y, Liu X, Tang X, Chen Q. Transcriptome Analysis of Berries of Spine Grape (Vitis davidii Föex) Infected by Colletotrichum viniferum during Symptom Development. Horticulturae. 2022; 8(9):843. https://doi.org/10.3390/horticulturae8090843
Chicago/Turabian StyleLei, Yan, Xiaojian Yuan, Ting Chen, Yuan Yuan, Xinming Liu, Xinbiao Tang, and Qingxi Chen. 2022. "Transcriptome Analysis of Berries of Spine Grape (Vitis davidii Föex) Infected by Colletotrichum viniferum during Symptom Development" Horticulturae 8, no. 9: 843. https://doi.org/10.3390/horticulturae8090843