
Systematic Identification and Validation of Housekeeping and Tissue-Specific Genes in Allotetraploid Chenopodium quinoa


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Isolation and cDNA Synthesis
2.3. RNA-Seq Analysis Pipeline
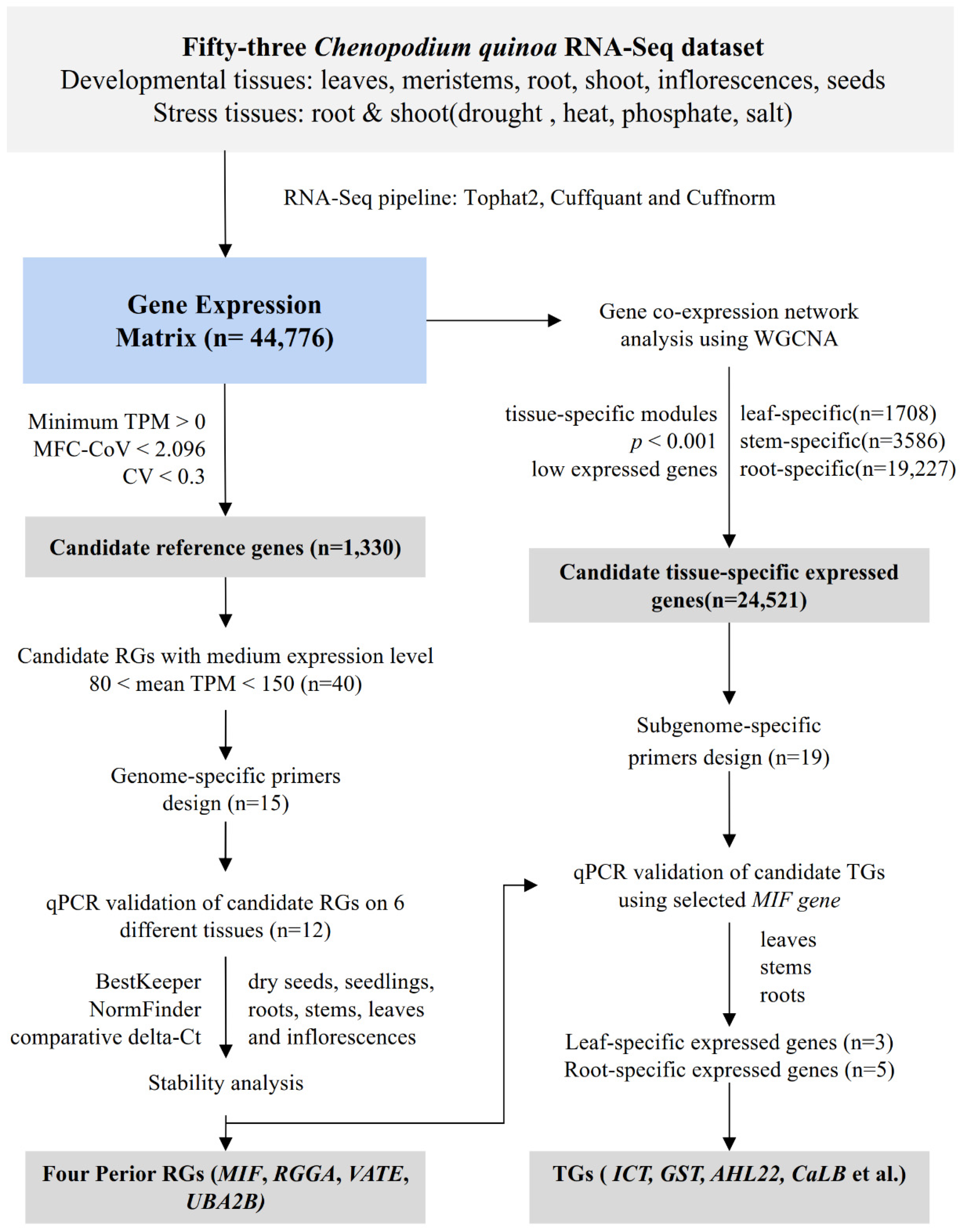
2.4. Systematic Identification of Housekeeping and Tissue-Specific Genes with RNA-Seq Data
2.5. qPCR Validation
2.6. Evaluation of Candidate Housekeeping Gene Stability
2.7. Gene Ontology Enrichment Analysis
3. Results
3.1. Expression Profiles of Candidate Housekeeping and Tissue-Specific Genes
3.2. qPCR Validation and Stability Measurement of Candidate Housekeeping Genes
3.3. qPCR Validation and Specificity Measurement of Candidate Tissue-Specific Genes
3.4. GO Enrichment of Housekeeping and Tissue-Specific Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adolf, V.I.; Shabala, S.; Andersen, M.N.; Razzaghi, F.; Jacobsen, S.E. Varietal differences of quinoa’s tolerance to saline conditions. Plant. Soil 2012, 357, 117–129. [Google Scholar] [CrossRef]
- Hariadi, Y.; Marandon, K.; Tian, Y.; Jacobsen, S.E.; Shabala, S. Ionic and osmotic relations in quinoa (Chenopodium quinoa Willd.) plants grown at various salinity levels. J. Exp. Bot. 2011, 62, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, S.E.; Mujica, A.; Jensen, C.R. The resistance of quinoa (Chenopodium quinoa Willd.) to adverse abiotic factors. Food Rev. Int. 2003, 19, 99–109. [Google Scholar] [CrossRef]
- Vega-Gálvez, A.; Miranda, M.; Vergara, J.; Uribe, E.; Puente, L.; Martínez, E.A. Nutrition facts and functional potential of quinoa (Chenopodium quinoa willd.), an ancient Andean grain: A review. J. Sci. Food Agric. 2010, 90, 2541–2547. [Google Scholar] [CrossRef]
- Jarvis, D.E.; Ho, Y.S.; Lightfoot, D.J.; Schmöckel, S.M.; Li, B.; Borm, T.J.A.; Ohyanagi, H.; Mineta, K.; Michell, C.T.; Saber, N.; et al. The genome of Chenopodium quinoa. Nature 2017, 542, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssens, N.; Janicot, M.; Perera, T.; Bakker, A. Housekeeping genes as internal standards in cancer research. Mol. Diagn. 2004, 8, 107–113. [Google Scholar] [CrossRef]
- de Jonge, H.J.M.; Fehrmann, R.S.N.; de Bont, E.S.J.M.; Hofstra, R.M.W.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.E.; van der Zee, A.G.J.; te Meerman, G.J.; ter Elst, A. Evidence based selection of housekeeping genes. PLoS ONE 2007, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Chari, R.; Lonergan, K.M.; Pikor, L.A.; Coe, B.P.; Zhu, C.Q.; Chan, T.H.; MacAulay, C.E.; Tsao, M.S.; Lam, S.; Ng, R.T.; et al. A sequence-based approach to identify reference genes for gene expression analysis. BMC Med. Genom. 2010, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, V.L.T.; Tom, L.N.; Quek, X.C.; Tan, J.M.; Payne, E.J.; Lin, L.L.; Sinnya, S.; Raphael, A.P.; Lambie, D.; Frazer, I.H.; et al. RNA-seq reveals more consistent reference genes for gene expression studies in human non-melanoma skin cancers. Peer J. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Dezsö, Z.; Nikolsky, Y.; Sviridov, E.; Shi, W.; Serebriyskaya, T.; Dosymbekov, D.; Bugrim, A.; Rakhmatulin, E.; Brennan, R.J.; Guryanov, A.; et al. A comprehensive functional analysis of tissue specificity of human gene expression. BMC Biol. 2008, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, L.L.; Dangond, F.; Yoshida, T.; Hong, R.; Jensen, R.V.; Misra, J.; Dillon, W.; Lee, K.F.; Clark, K.E.; Haverty, P.; et al. A compendium of gene expression in normal human tissues. Physiol. Genom. 2002, 2002, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.-S.; Jang, S.; Lee, S.; Nam, J.; Kim, C.; Lee, S.-H.; Chung, Y.-Y.; Kim, S.-R.; Lee, Y.H.; Cho, Y.-G.; et al. Leafy hull sterile1 is a Homeotic Mutation in a Rice MADS Box Gene Affecting Rice Flower Development. Plant. Cell 2000, 12, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Theißen, G. Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant. Biol. 2001, 4, 75–85. [Google Scholar] [CrossRef]
- Wu, H.; Gaur, U.; Mekchay, S.; Peng, X.; Li, L.; Sun, H.; Song, Z.; Dong, B.; Li, M.; Wimmers, K.; et al. Genome-wide identification of allele-specific expression in response to Streptococcus suis 2 infection in two differentially susceptible pig breeds. J. Appl. Genet. 2015, 56, 481–491. [Google Scholar] [CrossRef]
- Tan, H.; Yang, X.; Zhang, F.; Zheng, X.; Qu, C.; Mu, J.; Fu, F.; Li, J.; Guan, R.; Zhang, H.; et al. Enhanced seed oil production in canola by conditional expression of brassica napus LEAFY COTYLEDON1 and LEC1-LIKE in developing seeds. Plant. Physiol. 2011, 156, 1577–1588. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Zhou, F.; Lin, Y. Two novel positive cis-regulatory elements involved in green tissue-specific promoter activity in rice (Oryza sativa L ssp.). Plant. Cell Rep. 2012, 31, 1159–1172. [Google Scholar] [CrossRef]
- Geng, L.; Duan, X.; Liang, C.; Shu, C.; Song, F.; Zhang, J. Mining tissue-specific contigs from peanut (Arachis hypogaea L.) for promoter cloning by deep transcriptome sequencing. Plant. Cell Physiol. 2014, 55, 1793–1801. [Google Scholar] [CrossRef] [Green Version]
- Fiallos-Jurado, J.; Pollier, J.; Moses, T.; Arendt, P.; Barriga-Medina, N.; Morillo, E.; Arahana, V.; de Lourdes Torres, M.; Goossens, A.; Leon-Reyes, A. Saponin determination, expression analysis and functional characterization of saponin biosynthetic genes in Chenopodium quinoa leaves. Plant. Sci. 2016, 250, 188–197. [Google Scholar] [CrossRef]
- Morales, A.; Zurita-Silva, A.; Maldonado, J.; Silva, H. Transcriptional responses of chilean quinoa (Chenopodium quinoa Willd.) under water deficit conditions uncovers ABA-independent expression patterns. Front. Plant. Sci. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, K.B.; Maldonado, J.; Biondi, S.; Silva, H. RNA-seq analysis of salt-stressed versus non salt-stressed transcriptomes of chenopodium quinoa landrace R49. Genes 2019, 10, 1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, B.T.; Landry, J.R. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Fu, N.; Guo, S.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Chen, W.; Li, Y.; Zeng, R.; et al. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genom. 2009, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Patel, R.K.; Jhanwar, S.; Priya, P.; Bhattacharjee, A.; Yadav, G.; Bhatia, S.; Chattopadhyay, D.; Tyagi, A.K.; Jain, M. Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant. Physiol. 2011, 156, 1661–1678. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, M.S.; Kim, Y.M.; Yeom, S.I.; Cheong, K.; Kim, K.T.; Jeon, J.; Kim, S.; Kim, D.S.; Sohn, S.H.; et al. Integrative structural annotation of de novo RNA-Seq provides an accurate reference gene set of the enormous genome of the onion (Allium cepa L.). DNA Res. 2015, 22, 19–27. [Google Scholar] [CrossRef]
- Maldonado-Taipe, N.; Patirange, D.S.R.; Schmöckel, S.M.; Jung, C.; Emrani, N. Validation of suitable genes for normalization of diurnal gene expression studies in Chenopodium quinoa. PLoS ONE 2021, 16, e0233821. [Google Scholar] [CrossRef]
- Zou, C.; Chen, A.; Xiao, L.; Muller, H.M.; Ache, P.; Haberer, G.; Zhang, M.; Jia, W.; Deng, P.; Huang, R.; et al. A high-quality genome assembly of quinoa provides insights into the molecular basis of salt bladder-based salinity tolerance and the exceptional nutritional value. Cell Res. 2017, 27, 1327–1340. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000 Res. 2018, 7, 1338. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Z.; Huang, K.; Cai, C.; Cai, L.; Jiang, C.Y.; Feng, Y.; Liu, Z.; Zeng, Q.; Cheng, L.; Sun, Y.E.; et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 2013, 500, 593–597. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Hu, F.; Zhou, Y.; Wu, F.; Gaut, B.S. Maize transposable elements contribute to long non-coding RNAs that are regulatory hubs for abiotic stress response. BMC Genomics 2019, 20, 864. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W.; Tichopád, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Yanai, I.; Benjamin, H.; Shmoish, M.; Chalifa-Caspi, V.; Shklar, M.; Ophir, R.; Bar-Even, A.; Horn-Saban, S.; Safran, M.; Domany, E.; et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 2005, 21, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, G.; Wang, L.; Yu, C.; Li, M.; Song, S.; Hao, L.; Ma, L.; Zhang, Z. Computational identification and characterization of glioma candidate biomarkers through multi-omics integrative profiling. Biol. Direct 2020, 15, 1–14. [Google Scholar] [CrossRef]
- Beer, L.; Mlitz, V.; Gschwandtner, M.; Berger, T.; Narzt, M.S.; Gruber, F.; Brunner, P.M.; Tschachler, E.; Mildner, M. Bioinformatics approach for choosing the correct reference genes when studying gene expression in human keratinocytes. Exp. Dermatol. 2015, 24, 742–747. [Google Scholar] [CrossRef]
- Chang, C.W.; Cheng, W.C.; Chen, C.R.; Shu, W.Y.; Tsai, M.L.; Huang, C.L.; Hsu, I.C. Identification of human housekeeping genes and Tissue-Selective genes by microarray Meta-Analysis. PLoS ONE 2011, 6, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, A.W.T.; Shaw, G.T.W.; Hwang, M.J. Partitioning the human transcriptome using Hkera, a novel classifier of housekeeping and tissue-specific genes. PLoS ONE 2013, 8, e83040. [Google Scholar] [CrossRef] [PubMed]
- Bourquin, V.; Nishikubo, N.; Abe, H.; Brumer, H.; Denman, S.; Eklund, M.; Christiernin, M.; Teeri, T.T.; Sundberg, B.; Mellerowicz, E.J. Xyloglucan endotransglycosylases have a function during the formation of secondary cell walls of vascular tissues. Plant. Cell 2002, 14, 3073–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene_ID | Symbol | Gene Name | Forward Primer | Reverse Primer | Product (bp) |
|---|---|---|---|---|---|
| AUR62013045 | APX3 | L-ascorbate peroxidase 3 | TCGTCAACACAGAATACCT | GCACTCTTCCTCATTCCTA | 179 |
| AUR62038932 | AHRI | Ketol-acid reductoisomerase | GGTGTCTATGTTGCTCTAATG | AAGTCTTGCTGTGGTTGA | 174 |
| AUR62019540 | MDH1 | Malate dehydrogenase 1 | GGTTCAGCAACATTGTCTAT | TCTTCCACTCCATTCTTCC | 170 |
| AUR62020291 | RSZ22 | Serine/arginine-rich splicing factor RSZ22 | TGCTATGAGTGTGGTGAG | GGCTCCTCCTGTATCTTG | 109 |
| AUR62013759 | RGGA | RGG repeats nuclear RNA binding protein A-like | AAGTCTGTCAGCATTAACG | TTACCTCCACCACCATATC | 113 |
| AUR62010794 | VATE | V-type proton ATPase subunit E | GTTGCCGAAAGGATGATG | CCAGGAGGAAGATGAATAGT | 124 |
| AUR62030760 | MIF | Macrophage migration inhibitory factor homolog | CTTATGTTATGGTGGTGCTTA | TTGTTGGTGTCGGGATTA | 115 |
| AUR62009613 | ADF1 | Actin-depolymerizing factor 1 | GCCGTTATGCTGTGTATG | TGAATGCCATCCAACTCT | 160 |
| AUR62008267 | CYCL | Cytochrome c1-1 | CACATCTCATCCTCCTCTT | ATCTACCTCCAGCCATTATC | 134 |
| AUR62018470 | UBA2B | UBP1-associated protein 2B | TGTTCGTGTATAGGAGTGTT | CCTCTTGGCTGATGATGT | 145 |
| AUR62012065 | OEP163 | Outer envelope pore protein 16-3 | ACCAGGATTGATAAGGACTT | CCAACTGCTCCATTAACAA | 138 |
| AUR62032855 | HEX1 | Woronin body major protein | GCTTCCTATGACAAATGGG | CGTTCCGATTTCAATTCACT | 94 |
| AUR62042589 | GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | AGCAGCAGGTCCGTTGAAGG | ACCACCCGTTGGCTGTAACC | 180 |
| AUR62039382 | ACT1 | Actin-1 | CGTGTGGCTCCAGAAGAGCA | CCTGTTGTACGTCCACTGGCA | 167 |
| Gene_ID | Symbol | τ | Tissue with the Highest Expression | Maximum Relative Expression Level |
|---|---|---|---|---|
| AUR62005063 | L1 | 0.488 | Leaf | 1 |
| AUR62005589 | L2 | 0.968 | Leaf | 35.334 |
| AUR62005590 | L3 | 0.975 | Leaf | 35.09 |
| AUR62039526 | L4 | 0.94 | Leaf | 53.557 |
| AUR62034418 | L5 | 0.695 | Leaf | 2.193 |
| AUR62039803 | L6 | 0.69 | Leaf | 4.237 |
| AUR62033160 | R1 | 0.90 | Root | 9.51 |
| AUR62010528 | R2 | 0.91 | Root | 25.28 |
| AUR62016467 | R3 | 0.74 | Root | 4.72 |
| AUR62026665 | R4 | 0.62 | Root | 4.69 |
| AUR62038762 | R5 | 0.80 | Root | 16.22 |
| AUR62043174 | R6 | 0.92 | Root | 8.51 |
| AUR62002636 | R7 | 0.99 | Root | 32.67 |
| AUR62032910 | R8 | 0.89 | Root | 12.21 |
| AUR62009323 | R9 | 0.98 | Root | 35.02 |
| AUR62002145 | S1 | 0.65 | Stem | 2.00 |
| AUR62003648 | S2 | 0.39 | Root | 35.75 |
| AUR62028094 | S3 | 0.62 | Stem | 60.55 |
| AUR62017190 | S4 | 0.67 | Stem | 40.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, B.; Chen, H.; Shi, P.; Hu, F.; Song, W.; Meng, L.; Lv, Y. Systematic Identification and Validation of Housekeeping and Tissue-Specific Genes in Allotetraploid Chenopodium quinoa. Horticulturae 2021, 7, 235. https://doi.org/10.3390/horticulturae7080235
He B, Chen H, Shi P, Hu F, Song W, Meng L, Lv Y. Systematic Identification and Validation of Housekeeping and Tissue-Specific Genes in Allotetraploid Chenopodium quinoa. Horticulturae. 2021; 7(8):235. https://doi.org/10.3390/horticulturae7080235
Chicago/Turabian StyleHe, Bing, Hui Chen, Pibiao Shi, Fengqin Hu, Wenjing Song, Lin Meng, and Yuanda Lv. 2021. "Systematic Identification and Validation of Housekeeping and Tissue-Specific Genes in Allotetraploid Chenopodium quinoa" Horticulturae 7, no. 8: 235. https://doi.org/10.3390/horticulturae7080235