
Decoding the Genomic Landscape of Pomegranate: A Genome-Wide Analysis of Transposable Elements and Their Structural Proximity to Functional Genes
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Sequence Data for the Four Pomegranate Cultivars
2.2. Collection and Abundance Estimation of Full-Length Transposable Elements
2.3. Identification of Shared Transposable Element Insertion Sites and Phylogenetic Analysis
2.4. Localization of Shared Transposable Element Insertion Sites in Genes or Their Proximity
2.5. Profiling the Insertion Time of Full-Length LTR-Retrotransposons
2.6. Functional Analysis of Genes in Proximity to or Interrupted by Transposable Elements
3. Results
3.1. Collection and Estimation of Abundance of Full-Length Transposable Elements
3.2. Identification and Phylogenetic Analysis of Shared Transposable Element Insertion Sites
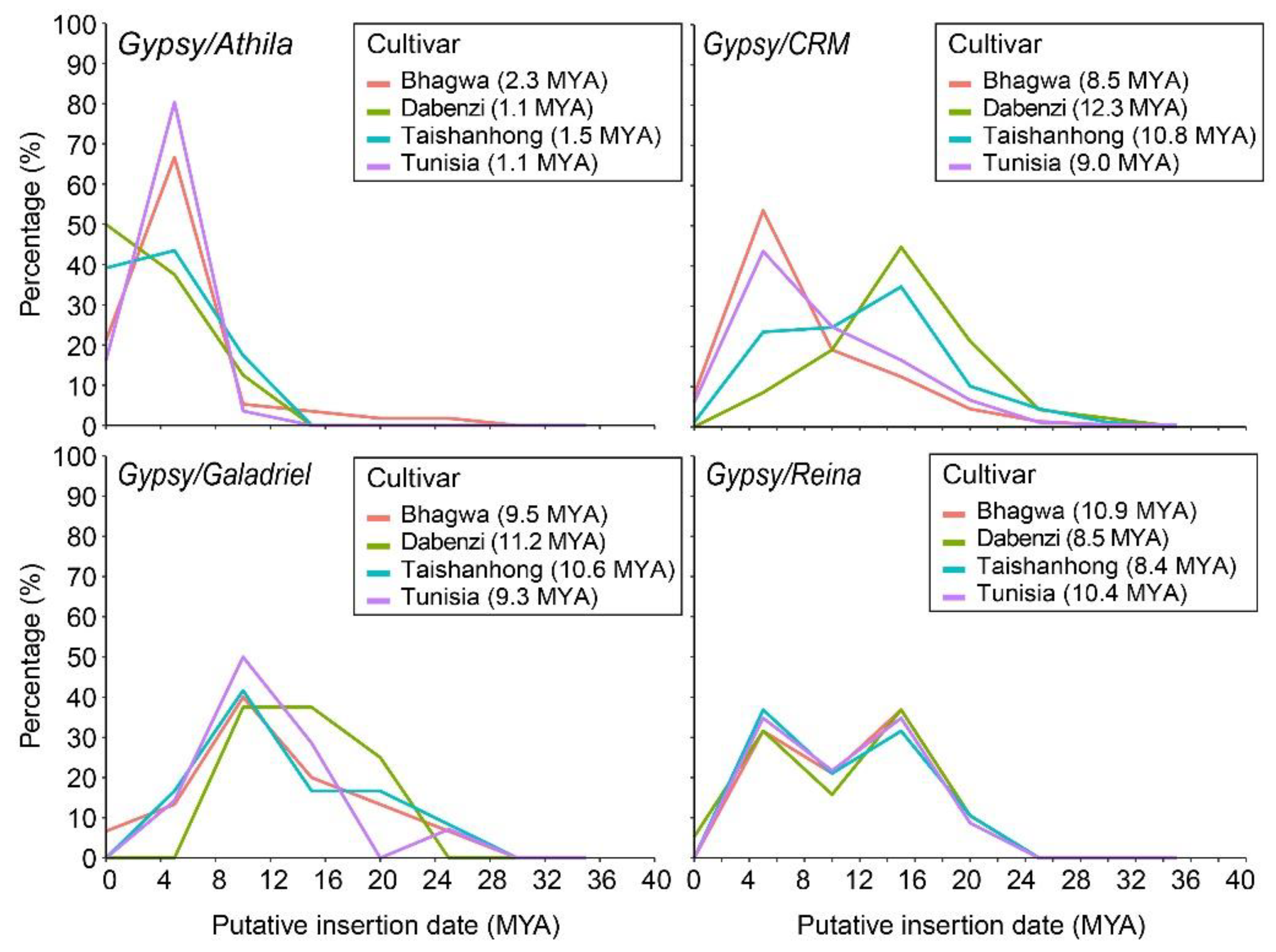
3.3. Profiling the Insertion Time of Full-Length LTR-Retrotransposons
3.4. Localization of Shared TE Insertion Sites in Genes or Their Proximity
3.5. Functional Analysis of Genes in Proximity to or Interrupted by Transposable Elements
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Patel, C.; Dadhaniya, P.; Hingorani, L.; Soni, M.G. Safety assessment of pomegranate fruit extract: Acute and subchronic toxicity studies. Food Chem. Toxicol. 2008, 46, 2728–2735. [Google Scholar] [CrossRef] [PubMed]
- Johanningsmeier, S.D.; Harris, G.K. Pomegranate as a functional food and nutraceutical source. Annu. Rev. Food Sci. Technol. 2011, 2, 181–201. [Google Scholar] [CrossRef]
- Sarkhosh, A.; Yavari, A.M.; Zamani, Z. (Eds.) The Pomegranate: Botany, Production and Uses; CABI: Wallingford, UK, 2020. [Google Scholar]
- Luo, X.; Li, H.; Wu, Z.; Yao, W.; Zhao, P.; Cao, D.; Yu, H.; Li, K.; Poudel, K.; Zhao, D.; et al. The pomegranate (Punica granatum L.) draft genome dissects genetic divergence between soft-and hard-seeded cultivars. Plant Biotechnol. J. 2020, 18, 955–968. [Google Scholar] [CrossRef]
- Qin, G.; Xu, C.; Ming, R.; Tang, H.; Guyot, R.; Kramer, E.M.; Hu, Y.; Yi, X.; Qi, Y.; Xu, X. The pomegranate (Punica granatum L.) genome and the genomics of punicalagin biosynthesis. Plant J. 2017, 91, 1108–1128. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Fang, Y.; Zhang, T.; Fei, Z.; Han, F.; Liu, C.; Liu, M.; Xiao, W.; Zhang, W.; Wu, S.; et al. The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol. J. 2018, 16, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Sowjanya, P.R.; Shilpa, P.; Patil, G.P.; Babu, D.K.; Sharma, J.; Sangnure, V.R.; Mundewadikar, D.M.; Natarajan, P.; Marathe, A.R.; Reddy, U.K.; et al. Reference quality genome sequence of Indian pomegranate cv.’Bhagawa’ (Punica granatum L.). Front. Plant Sci. 2022, 13, 947164. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef]
- Mascagni, F.; Barghini, E.; Giordani, T.; Rieseberg, L.H.; Cavallini, A.; Natali, L. Repetitive DNA and plant domestication: Variation in copy number and proximity to genes of LTR-retrotransposons among wild and cultivated sunflower (Helianthus annuus) genotypes. Genome Biol. Evol. 2015, 7, 3368–3382. [Google Scholar] [CrossRef]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramirez-Gonzalez, R.H.; De Oliveira, R. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.J.; Blomberg, J.; Coffin, J.M.; Fan, H.; Heidmann, T.; Mayer, J.; Stoye, J.; Tristem, M.; Johnson, W.E. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 2018, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Presting, G.G. Centromeric retrotransposon lineages predate the maize/rice divergence and differ in abundance and activity. Mol. Genet. Genom. 2008, 279, 133–147. [Google Scholar] [CrossRef]
- Gong, Z.; Wu, Y.; Koblížková, A.; Torres, G.A.; Wang, K.; Iovene, M.; Neumann, P.; Zhang, W.; Novák, P.; Buell, C.R.; et al. Repeatless and repeat-based centromeres in potato: Implications for centromere evolution. Plant Cell 2012, 24, 3559–3574. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Liu, Y.; Liu, Y.-X.; Lv, Z.; Li, H.; Xie, S.; Gao, Z.; Pang, J.; Wang, X.-J.; Lai, J.; et al. Dynamic chromatin changes associated with de novo centromere formation in maize euchromatin. Plant J. 2016, 88, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Neumann, P.; Navrátilová, A.; Koblížková, A.; Kejnovský, E.; Hřibová, E.; Hobza, R.; Widmer, A.; Doležel, J.; Macas, J. Plant centromeric retrotransposons: A structural and cytogenetic perspective. Mob. DNA 2011, 2, 4. [Google Scholar] [CrossRef]
- Muñoz-López, M.; García-Pérez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef]
- Morgante, M.; Brunner, S.; Pea, G.; Fengler, K.; Zuccolo, A.; Rafalski, A. Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat. Genet. 2005, 37, 997–1002. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Viviani, A.; Ventimiglia, M.; Fambrini, M.; Vangelisti, A.; Mascagni, F.; Pugliesi, C.; Usai, G. Impact of transposable elements on the evolution of complex living systems and their epigenetic control. Biosystems 2021, 210, 104566. [Google Scholar] [CrossRef] [PubMed]
- Devos, K.M.; Brown, J.K.; Bennetzen, J.L. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002, 12, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Panaud, O. Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol. Biol. Evol. 2003, 20, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dooner, H.K. Remarkable variation in maize genome structure inferred from haplotype diversity at the bz locus. Proc. Natl. Acad. Sci. USA 2006, 103, 17644–17649. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.R.; McComb, J.A.; Greene, W.K. Transposable elements: Powerful contributors to angiosperm evolution and diversity. Genome Biol. Evol. 2013, 5, 1886–1901. [Google Scholar] [CrossRef]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef]
- Pinosio, S.; Giacomello, S.; Faivre-Rampant, P.; Taylor, G.; Jorge, V.; Le Paslier, M.C.; Morgante, M. Characterization of the poplar pan-genome by genome-wide identification of structural variation. Mol. Biol. Evol. 2016, 33, 2706–2719. [Google Scholar] [CrossRef]
- Ventimiglia, M.; Marturano, G.; Vangelisti, A.; Usai, G.; Simoni, S.; Cavallini, A.; Giordani, T.; Natali, L.; Zuccolo, A.; Mascagni, F. Genome wide identification and characterization of exapted transposable elements in the large genome of sunflower (Helianthus annuus L.). Plant J. 2023, 113, 734–748. [Google Scholar] [CrossRef]
- Hirsch, C.D.; Springer, N.M. Transposable element influences on gene expression in plants. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 157–165. [Google Scholar] [CrossRef]
- Drongitis, D.; Aniello, F.; Fucci, L.; Donizetti, A. Roles of transposable elements in the different layers of gene expression regulation. Int. J. Mol. Sci. 2019, 20, 5755. [Google Scholar] [CrossRef] [PubMed]
- Fambrini, M.; Usai, G.; Vangelisti, A.; Mascagni, F.; Pugliesi, C. The plastic genome: The impact of transposable elements on gene functionality and genomic structural variations. Genesis 2020, 58, e23399. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, Y.; Liu, Z.; He, J.; Jiang, X.; He, F.; Lu, Z.; Yang, S.; Chen, P.; Yu, H.; et al. Somatic variations led to the selection of acidic and acidless orange cultivars. Nat. Plants 2021, 7, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.N.; Pei, M.S.; Ampomah-Dwamena, C.; He, G.Q.; Wei, T.L.; Shi, Q.F.; Yu, Y.H.; Guo, D.L. Genome-wide characterization of long terminal repeat retrotransposons provides insights into trait evolution of four cucurbit species. Funct. Integr. Genom. 2023, 23, 218. [Google Scholar] [CrossRef] [PubMed]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef] [PubMed]
- Dubin, M.J.; Scheid, O.M.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef]
- Schrader, L.; Schmitz, J. The impact of transposable elements in adaptive evolution. Mol. Ecol. 2019, 28, 1537–1549. [Google Scholar] [CrossRef]
- Vangelisti, A.; Simoni, S.; Usai, G.; Ventimiglia, M.; Natali, L.; Cavallini, A.; Mascagni, F.; Giordani, T. LTR-retrotransposon dynamics in common fig (Ficus carica L.) genome. BMC Plant Biol. 2021, 21, 221. [Google Scholar] [CrossRef]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 2004, 304, 982. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, J.; Han, X.; Li, J.; Gao, Y.; Richards, C.M.; Zhang, C.; Tian, Y.; Liu, G.; Gul, H.; et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 2019, 10, 1494. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Liang, C. Generic repeat finder: A high-sensitivity tool for genome-wide de novo repeat detection. Plant Physiol. 2019, 180, 1803–1815. [Google Scholar] [CrossRef]
- Su, W.; Gu, X.; Peterson, T. TIR-Learner, a new ensemble method for TIR transposable element annotation, provides evidence for abundant new transposable elements in the maize genome. Mol. Plant 2019, 12, 447–460. [Google Scholar] [CrossRef]
- Xiong, W.; He, L.; Lai, J.; Dooner, H.K.; Du, C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl. Acad. Sci. USA 2014, 111, 10263–10268. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 3 July 2023).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]
- Wickham, H. Data analysis. In ggplot2. Use R! Springer: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.A. simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Usai, G.; Natali, L.; Cavallini, A.; Giordani, T. A comparison of methods for LTR-retrotransposon insertion time profiling in the Populus trichocarpa genome. Caryologia 2018, 71, 85–92. [Google Scholar] [CrossRef]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Usai, G.; Mascagni, F.; Natali, L.; Giordani, T.; Cavallini, A. Comparative genome-wide analysis of repetitive DNA in the genus Populus L. Tree Genet. Genomes 2017, 13, 96. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, 7. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Z.; Shi, Z.; Zhang, S.; Ming, R.; Zhu, S.; Khan, M.A.; Tao, S.; Korban, S.S.; Wang, H.; et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013, 23, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Usai, G.; Mascagni, F.; Giordani, T.; Vangelisti, A.; Bosi, E.; Zuccolo, A.; Ceccarelli, M.; King, R.; Hassani-Pak, K.; Liceth, S.Z.; et al. Epigenetic patterns within the haplotype phased fig (Ficus carica L.) genome. Plant J. 2020, 102, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Aryal, R.; Dudchenko, O.; Sargent, D.J.; Mead, D.; Buti, M.; Cavallini, A.; Hytönen, T.; Andrés, J.; Pham, M.; et al. A chromosome-length genome assembly and annotation of blackberry (Rubus argutus, cv. “Hillquist”). G3 2023, 13, jkac289. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Koblížková, A.; Navrátilová, A.; Macas, J. Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics 2006, 173, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Christelová, P.; Valárik, M.; Hřibová, E.; De Langhe, E.; Doležel, J. A multi gene sequence-based phylogeny of the Musaceae (banana) family. BMC Evol. Biol. 2011, 11, 130. [Google Scholar] [CrossRef]
- Tenaillon, M.I.; Hufford, M.B.; Gaut, B.S.; Ross-Ibarra, J. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol. Evol. 2011, 3, 219–229. [Google Scholar] [CrossRef]
- Vitte, C.; Fustier, M.A.; Alix, K.; Tenaillon, M.I. The bright side of transposons in crop evolution. Brief. Funct. Genom. 2014, 13, 276–295. [Google Scholar] [CrossRef]
- Kreplak, J.; Madoui, M.A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Burstin, J. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Macas, J.; Novák, P.; Pellicer, J.; Čížková, J.; Koblížková, A.; Neumann, P.; Leitch, I.J. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE 2015, 10, e0143424. [Google Scholar] [CrossRef]
- Simoni, S.; Clemente, C.; Usai, G.; Vangelisti, A.; Natali, L.; Tavarini, S.; Angelini, C.L.; Cavallini, A.; Mascagni, F.; Giordani, T. Characterisation of LTR-retrotransposons of Stevia rebaudiana and their use for the analysis of genetic variability. Int. J. Mol. Sci. 2022, 23, 6220. [Google Scholar] [CrossRef]
- He, G.Q.; Jin, H.Y.; Cheng, Y.Z.; Yu, Y.H.; Guo, D.L. Characterization of genome-wide long terminal repeat retrotransposons provide insights into trait evolution of four grapevine species. J. Syst. Evol. 2023, 61, 414–427. [Google Scholar] [CrossRef]
- Usai, G.; Mascagni, F.; Vangelisti, A.; Giordani, T.; Ceccarelli, M.; Cavallini, A.; Natali, L. Interspecific hybridisation and LTR-retrotransposon mobilisation-related structural variation in plants: A case study. Genomics 2020, 112, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.J.; Gao, L.Z. Rapid and recent evolution of LTR retrotransposons drives rice genome evolution during the speciation of AA-genome Oryza species. G3 2017, 7, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, P.; Goubely, C.; Pelissier, T.; Deragon, J.M. SINE retroposons can be used in vivo as nucleation centers for de novo methylation. Mol. Cell Biol. 2000, 20, 3434–3441. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.I.; Tomás-Barberán, F.A.; Hess-Pierce, B.; Holcroft, D.M.; Kader, A.A. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J. Agric. Food Chem. 2000, 48, 4581–4589. [Google Scholar] [CrossRef] [PubMed]
- Longtin, R. The pomegranate: Nature’s power fruit? J. Natl. Cancer Inst. 2003, 95, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Tzulker, R.; Glazer, I.; Bar-Ilan, I.; Holland, D.; Aviram, M.; Amir, R. Antioxidant activity, polyphenol content, and related compounds in different fruit juices and homogenates prepared from 29 different pomegranate accessions. J. Agric. Food Chem. 2007, 55, 9559–9570. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, S.; Sithul, H.; Muraleedharan, P.; Azeez, J.M.; Sreeharshan, S. Pomegranate fruit as a rich source of biologically active compounds. Biomed. Res. Int. 2014, 2014, 686921. [Google Scholar] [CrossRef]
- Díaz-Mula, H.M.; Tomás-Barberán, F.A.; García-Villalba, R. Pomegranate fruit and juice (cv. Mollar), rich in ellagitannins and anthocyanins, also provide a significant content of a wide range of proanthocyanidins. J. Agric. Food Chem. 2019, 67, 9160–9167. [Google Scholar] [CrossRef]
- Horbowicz, M.; Kosson, R.; Grzesiuk, A.; Dębski, H. Anthocyanins of fruits and vegetables-their occurrence, analysis and role in human nutrition. J. Fruit Ornam. Plant Res. 2008, 68, 5–22. [Google Scholar] [CrossRef]
- Zhao, J.; Qi, X.; Li, J.; Cao, Z.; Liu, X.; Yu, Q.; Qin, G. Metabolic Profiles of Pomegranate Juices during Fruit Development and the Redirection of Flavonoid Metabolism. Horticulturae 2023, 9, 881. [Google Scholar] [CrossRef]
- Zhu, F.; Yuan, Z.; Zhao, X.; Yin, Y.; Feng, L. Composition and contents of anthocyanins in different pomegranate cultivars. Acta Hortic 2015, 1089, 35–41. [Google Scholar]
- Lavhale, S.G.; Kalunke, R.M.; Giri, A.P. Structural, functional and evolutionary diversity of 4-coumarate-CoA ligase in plants. Planta 2018, 248, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jiang, X.L.; Qian, Y.M.; Wang, P.Q.; Xie, D.Y.; Gao, L.P.; Xia, T. Metabolic characterization of the anthocyanidin reductase pathway involved in the biosynthesis of Flavan-3-ols in elite Shuchazao Tea (Camellia sinensis) cultivar in the field. Molecules 2017, 22, 2241. [Google Scholar] [CrossRef]
- Wisman, E.; Hartmann, U.; Sagasser, M.; Baumann, E.; Palme, K.; Hahlbrock, K.; Saedler, H.; Weisshaar, B. Knock-out mutants from an En-1 mutagenized Arabidopsis thaliana population generate phenylpropanoid biosynthesis phenotypes. Proc. Natl. Acad. Sci. USA 1998, 95, 12432–12437. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Superfamily | Lineage | Tunisia (nr) | Bhagwa (nr) | Taishanhong (nr) | Dabenzi (nr) |
|---|---|---|---|---|---|---|
| Class I (Retrotransposons) | Copia | Ale | 179 | 179 | 148 | 132 |
| Alesia | 1 | 1 | 1 | 1 | ||
| Angela | 229 | 230 | 70 | 20 | ||
| Ikeros | 10 | 10 | 10 | 7 | ||
| Ivana | 53 | 52 | 32 | 32 | ||
| TAR | 66 | 65 | 30 | 29 | ||
| Tork | 148 | 143 | 71 | 23 | ||
| Gypsy | Chromovirus/CRM | 190 | 229 | 90 | 46 | |
| Chromovirus/Galadriel | 14 | 13 | 12 | 8 | ||
| Chromovirus/Reina | 24 | 24 | 19 | 19 | ||
| Chromovirus/Tekay | 6 | 8 | 1 | 1 | ||
| Non-Chromovirus/Athila | 56 | 52 | 23 | 8 | ||
| Non-Chromovirus/Tat/Ogre | 59 | 58 | 8 | 1 | ||
| Unknown | 121 | 138 | 63 | 57 | ||
| LINE | 1 | 1 | nd | nd | ||
| Pararetrovirus | nd | nd | 1 | nd | ||
| Class II (DNA Transposons) | TIR | hAT | 127 | 110 | 111 | 110 |
| CACTA | 141 | 171 | 148 | 142 | ||
| PIF/Harbinger | 28 | 37 | 27 | 29 | ||
| Mutator | 393 | 374 | 368 | 356 | ||
| Tc1/Mariner | 19 | 17 | 13 | 13 | ||
| MITE | hAT | 88 | 90 | 84 | 78 | |
| CACTA | 15 | 15 | 12 | 16 | ||
| PIF/Harbinger | 16 | 12 | 15 | 14 | ||
| Mutator | 96 | 96 | 85 | 90 | ||
| Tc1/Mariner | 1 | 2 | 4 | 5 | ||
| Helitron | Helitron | 378 | 373 | 371 | 366 | |
| Unknown | 6 | 11 | 5 | 3 | ||
| Total | 2465 | 2511 | 1822 | 1606 |
| Order | Superfamily | Lineage | Tunisia (%) | Bhagwa (%) | Taishanhong (%) | Dabenzi (%) |
|---|---|---|---|---|---|---|
| Class I (Retrotransposons) | Copia | Ale | 1.72 | 1.61 | 1.79 | 1.80 |
| Alesia | 0.01 | 0.01 | 0.01 | 0.01 | ||
| Angela | 2.26 | 2.08 | 1.49 | 1.90 | ||
| Ikeros | 0.15 | 0.13 | 0.15 | 0.16 | ||
| Ivana | 0.33 | 0.32 | 0.33 | 0.36 | ||
| TAR | 0.44 | 0.41 | 0.34 | 0.4 | ||
| Tork | 0.72 | 0.7 | 0.56 | 0.65 | ||
| Total | 5.63 | 5.26 | 4.67 | 5.28 | ||
| Gypsy | Chromovirus/CRM | 3.05 | 3.22 | 1.65 | 2.15 | |
| Chromovirus/Galadriel | 0.08 | 0.07 | 0.08 | 0.08 | ||
| Chromovirus/Reina | 0.12 | 0.11 | 0.13 | 0.13 | ||
| Chromovirus/Tekay | 0.48 | 0.62 | 0.18 | 0.26 | ||
| Non-Chromovirus/Athila | 0.48 | 0.45 | 0.36 | 0.4 | ||
| Non-Chromovirus/Tat/Ogre | 8.88 | 8.98 | 4.98 | 6.59 | ||
| Total | 13.09 | 13.45 | 7.38 | 9.61 | ||
| Total Copia/Gypsy | 18.72 | 18.71 | 12.05 | 14.89 | ||
| Unknown | 11.88 | 15.32 | 7.42 | 9.0 | ||
| LINE | 0.05 | 0.05 | 0.06 | 0.05 | ||
| pararetrovirus | 0.08 | 0.07 | 0.08 | 0.08 | ||
| Class II (DNA Transposons) | TIR | hAT | 1.32 | 1.08 | 1.5 | 1.23 |
| CACTA | 3.13 | 2.93 | 3.71 | 3.32 | ||
| PIF/Harbinger | 1.26 | 1.19 | 1.42 | 1.35 | ||
| Mutator | 5.61 | 5.27 | 6.27 | 5.99 | ||
| Tc1/Mariner | 0.38 | 0.36 | 0.43 | 0.41 | ||
| MITE | hAT | 0.24 | 0.27 | 0.36 | 0.3 | |
| CACTA | 0.04 | 0.04 | 0.05 | 0.05 | ||
| PIF/Harbinger | 0.07 | 0.07 | 0.11 | 0.08 | ||
| Mutator | 0.42 | 0.39 | 0.55 | 0.46 | ||
| Tc1/Mariner | 0.02 | 0.02 | 0.05 | 0.02 | ||
| Helitron | Helitron | 6.9 | 6.68 | 7.61 | 7.45 | |
| Total | 50.12 | 52.45 | 41.67 | 44.68 |
| Genotype Combination | Number of Insertion Sites (nr) | ||
|---|---|---|---|
| Total Transposable Elements | Retrotransposons | DNA Transposons | |
| Bhagwa–Dabenzi–Taishanhong–Tunisia | 73 | 23 | 50 |
| Bhagwa–Dabenzi–Taishanhong | 200 | 38 | 162 |
| Bhagwa–Dabenzi–Tunisia | 81 | 17 | 64 |
| Bhagwa–Taishanhong–Tunisia | 125 | 63 | 62 |
| Dabenzi–Taishanhong–Tunisia | 211 | 38 | 173 |
| Bhagwa–Dabenzi | 310 | 68 | 242 |
| Bhagwa–Taishanhong | 362 | 125 | 237 |
| Bhagwa–Tunisia | 402 | 252 | 150 |
| Dabenzi–Taishanhong | 137 | 30 | 107 |
| Dabenzi–Tunisia | 293 | 68 | 225 |
| Taishanhong–Tunisia | 396 | 141 | 255 |
| Bhagwa | 947 | 621 | 326 |
| Dabenzi | 297 | 105 | 192 |
| Taishanhong | 313 | 124 | 189 |
| Tunisia | 878 | 554 | 324 |
| Cultivar | TE Insertion Location | ||
|---|---|---|---|
| Close to Gene (nr) | Exon (nr) | Intron (nr) | |
| ‘Bhagwa’ | 1264 | 66 | 69 |
| ‘Dabenzi’ | 874 | 40 | 50 |
| ‘Taishanhong’ | 968 | 39 | 58 |
| ‘Tunisia’ | 1278 | 61 | 63 |
| Source of variation | Percentage of variation (%) | Significance | |
| Cultivar | 1.07 | ns | |
| TE insertion location | 95.76 | *** | |
| Insertion Location | Transposable Element Family/Lineage | Gene ID | Gene Name | Gene Code | Genotype Combination |
|---|---|---|---|---|---|
| Close to gene | Copia/Ale | XM_031520924.1 | flavonoid 3′-monooxygenase | F3′H | Bh|Da|Ta|Tu |
| Copia/Ivana | XM_031528957.1 | flavonoid 3′-monooxygenase | F3′H | Bh|Da|Ta|Tu | |
| Gypsy/Chromovirus/CRM | XM_031526933.1 | 4-coumarate--CoA ligase | 4CL | Bh|Ta | |
| Helitron | XM_031516428.1 | 4-coumarate--CoA ligase | 4CL | Da|Ta|Tu | |
| Exon | TIR/Mariner | XM_031530037.1 | anthocyanidin reductase | ANR | Tu |
| Gypsy/Chromovirus/Reina | XM_031520605.1 | peroxidase | POD | Bh|Da | |
| Gypsy/Chromovirus/Reina | XM_031520605.1 | peroxidase | POD | Ta|Tu |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simoni, S.; Usai, G.; Vangelisti, A.; Castellacci, M.; Giordani, T.; Natali, L.; Mascagni, F.; Cavallini, A. Decoding the Genomic Landscape of Pomegranate: A Genome-Wide Analysis of Transposable Elements and Their Structural Proximity to Functional Genes. Horticulturae 2024, 10, 111. https://doi.org/10.3390/horticulturae10020111
Simoni S, Usai G, Vangelisti A, Castellacci M, Giordani T, Natali L, Mascagni F, Cavallini A. Decoding the Genomic Landscape of Pomegranate: A Genome-Wide Analysis of Transposable Elements and Their Structural Proximity to Functional Genes. Horticulturae. 2024; 10(2):111. https://doi.org/10.3390/horticulturae10020111
Chicago/Turabian StyleSimoni, Samuel, Gabriele Usai, Alberto Vangelisti, Marco Castellacci, Tommaso Giordani, Lucia Natali, Flavia Mascagni, and Andrea Cavallini. 2024. "Decoding the Genomic Landscape of Pomegranate: A Genome-Wide Analysis of Transposable Elements and Their Structural Proximity to Functional Genes" Horticulturae 10, no. 2: 111. https://doi.org/10.3390/horticulturae10020111