
Integrated Metagenomics and Network Analysis of Metabolic Functional Genes in the Microbial Community of Chinese Fermentation Pits
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
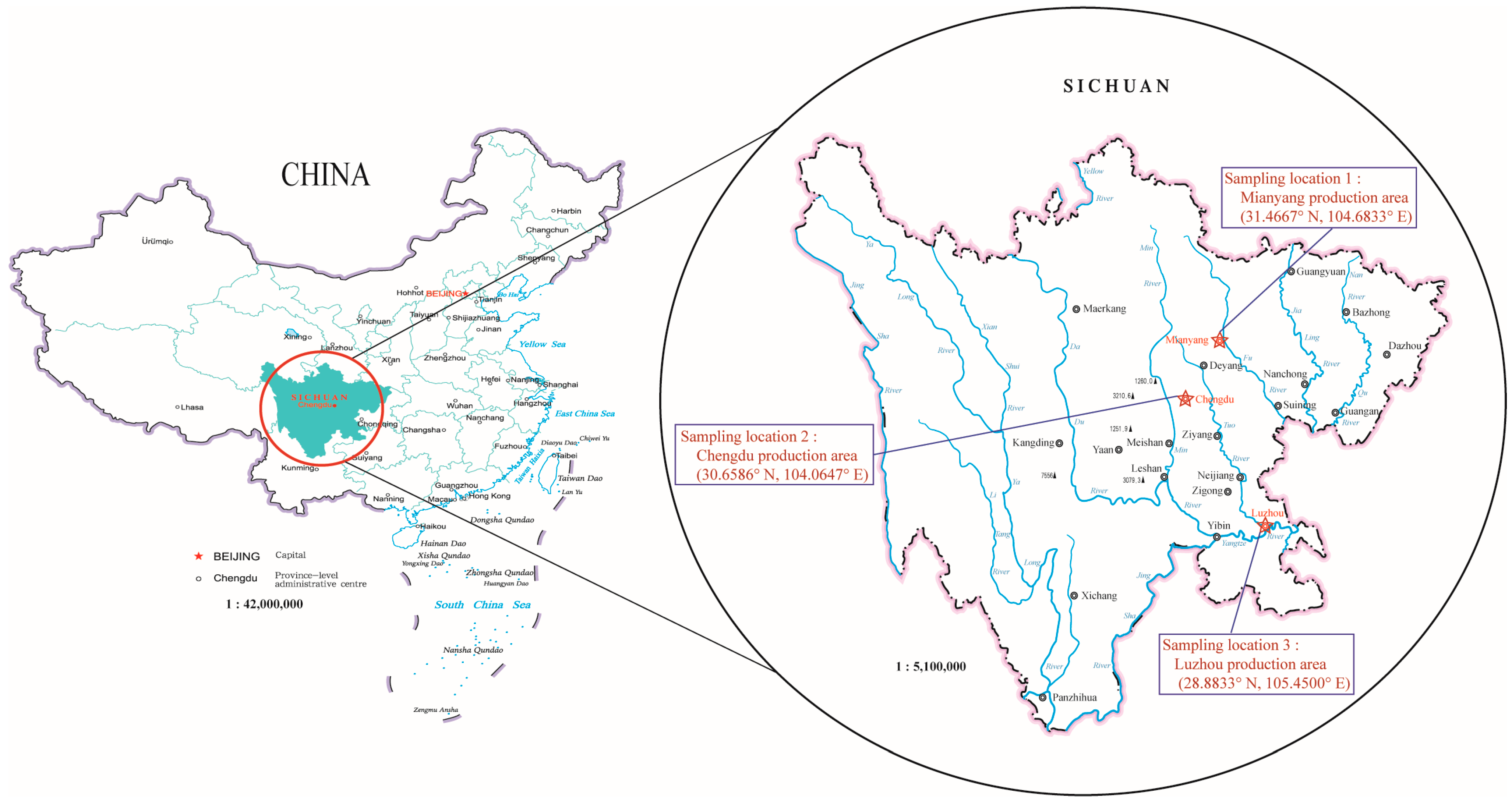
2.1. Sampling and Metagenomic DNA Extraction
2.2. Paired-End (PE) Library Generation and Illumina Hiseq Sequencing
2.3. Data Processing of Metagenomic Sequence Reads
2.4. Metagenome Assembly of Fps
2.5. Gene Prediction and Abundance Analysis in FPs
2.6. Gene Functional Annotation of FPs Aligned to the KEGG/eggNOG/CAZy Databases
2.7. Drawing the Overall Metabolic Pathway Profile of FPs
2.8. Construction of the Generative Pathways for the Main Flavor Compounds of CSAB
2.9. Statistical Analysis
3. Results and Discussion
3.1. Statistical Analysis of Fundamental Information for FPs Metagenomic Data
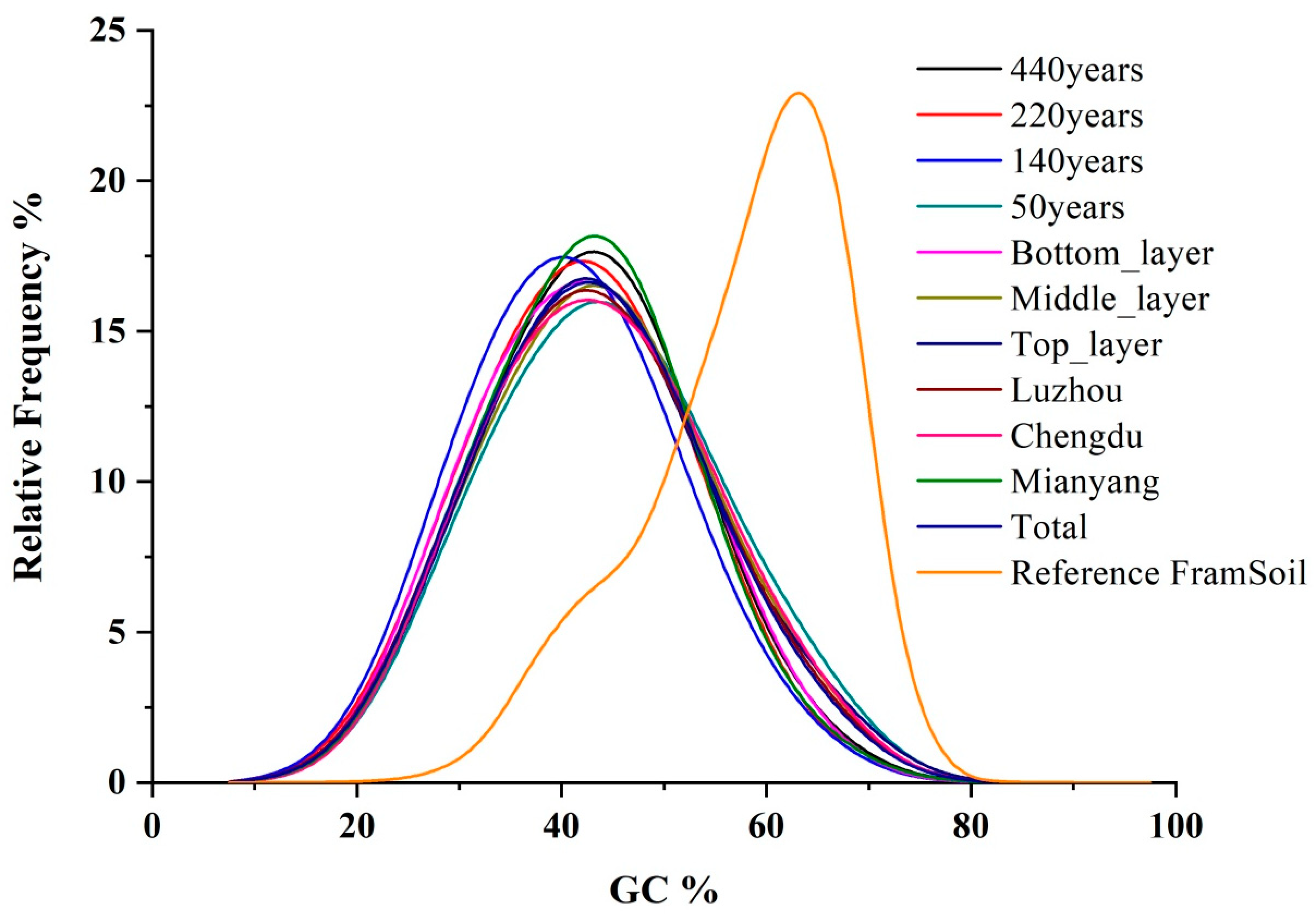
3.2. Assembly Results of FPs Metagenome
3.3. Gene Prediction Characteristics of FPs
3.4. Comparative and Differential Analysis of Gene Sets in FPs
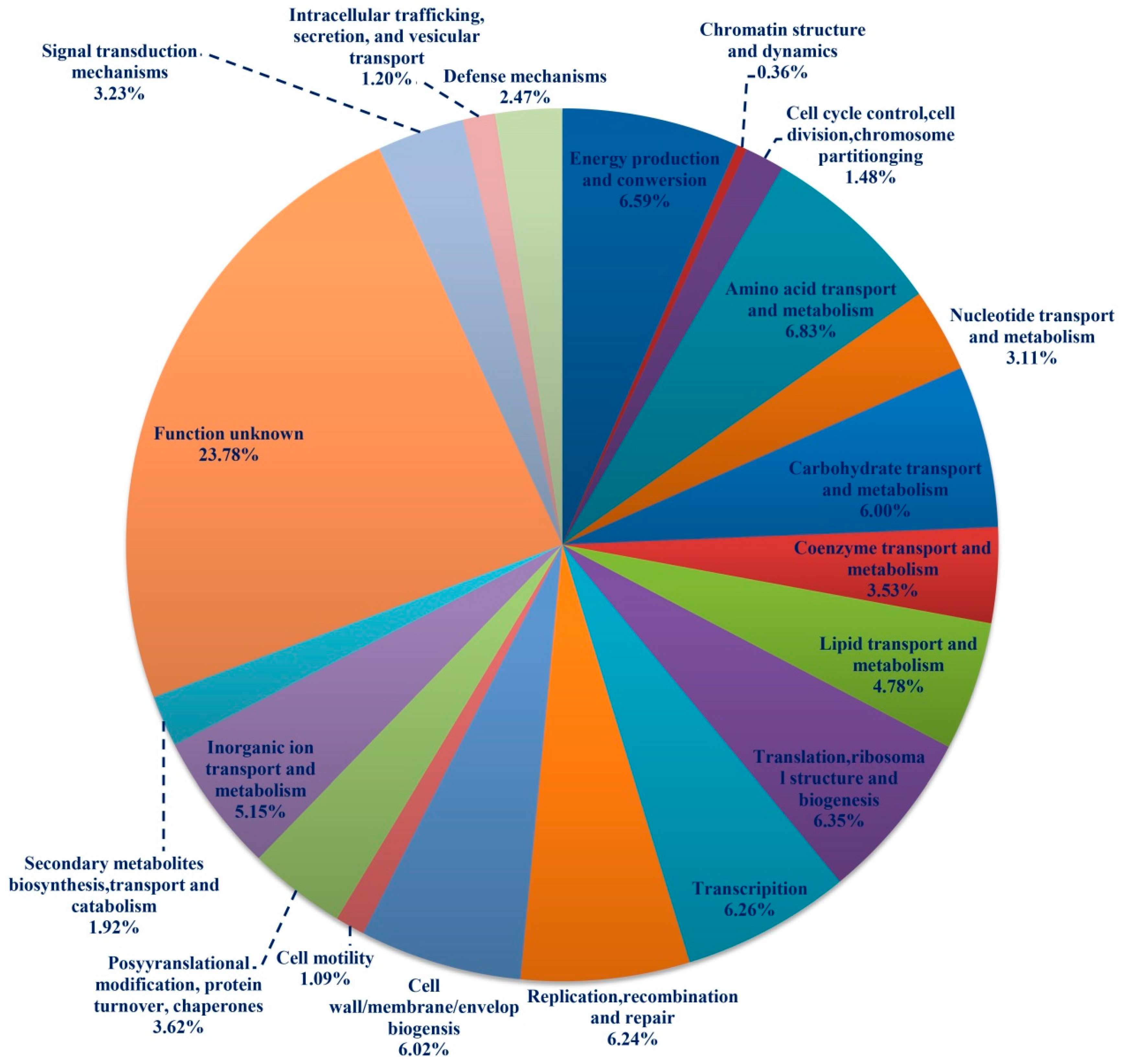
3.5. Classificatory and Differential Analysis of Functional Genes in FPs
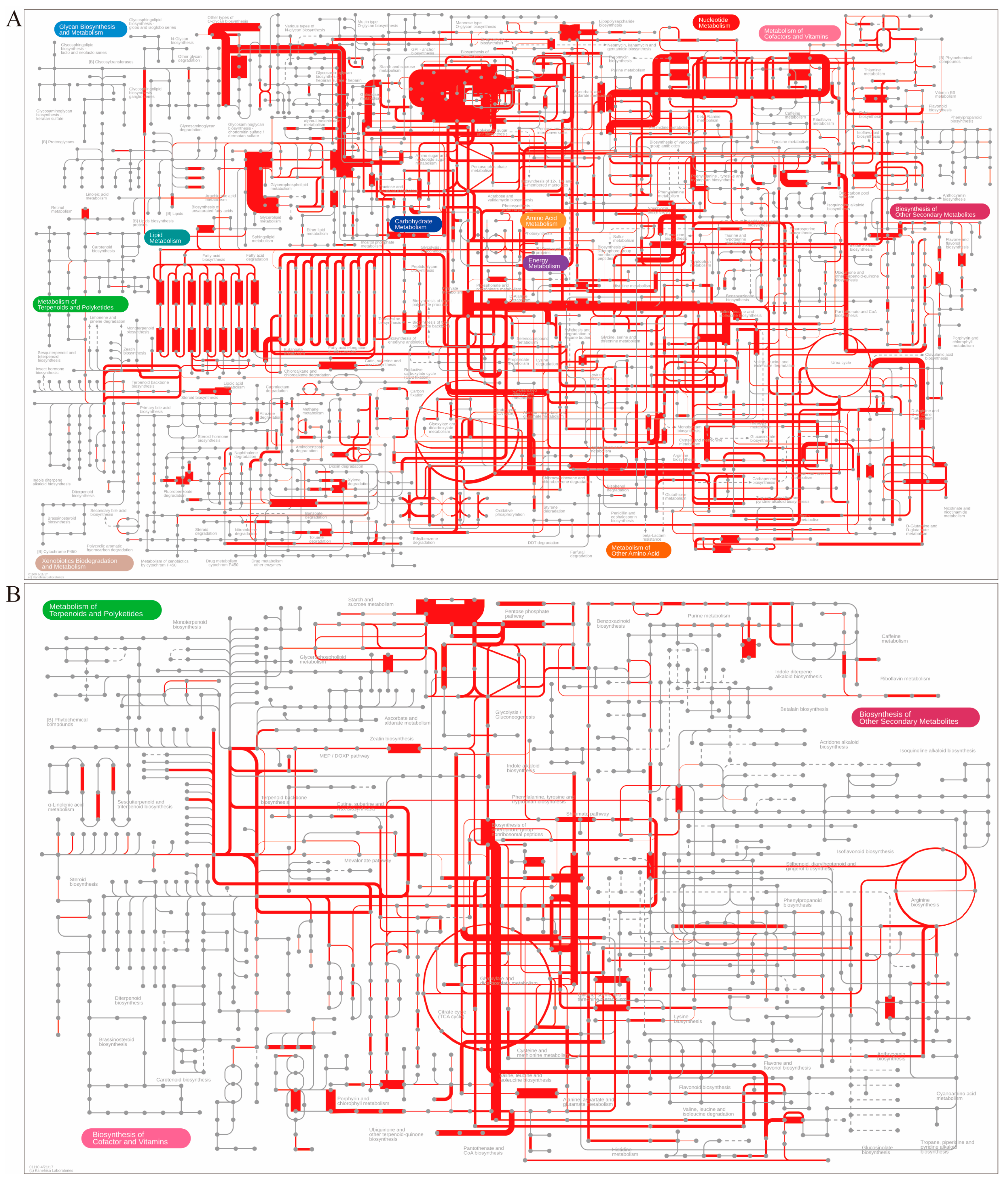
3.6. Overall Characteristics of the Constructed Metabolic Pathway Profile of FPs
3.7. The Deciphered Generative Pathway Profile for the Main Flavor Compounds of CSAB
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAs | Auxiliary activities |
| ANOVA | Analysis of variance |
| BLAST | Basic local alignment search tool |
| CAZy | Carbohydrate active enzyme |
| CBMs | Carbohydrate-binding modules |
| CEs | Carbohydrate esterases |
| CoA | Coenzyme A |
| COG | Clusters of orthologous genes |
| CSAB | Chinese strong-aroma baijiu |
| eggNOG | Evolutionary gene genealogy non-supervised orthologous groups |
| FP | Fermentation pit |
| GC-MS | Gas chromatography-mass spectrometry |
| GHs | Glycoside hydrolases |
| GTs | Glycosyl transferases |
| HMM | Hidden Markov model |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KO | KEGG orthology |
| LC-MS | Liquid chromatography-mass spectrometry |
| MCL | Markov cluster algorithm |
| NAD | Nicotinamide adenine dinucleotide |
| OGs | Orthologous groups |
| ORFs | Open reading frames |
| PE | Paired-end |
| PLs | Polysaccharide lyases |
References
- Ye, H.; Wang, J.; Shi, J.; Du, J.; Zhou, Y.; Huang, M.; Sun, B. Automatic and Intelligent Technologies of Solid-State Fermentation Process of Baijiu Production: Applications, Challenges, and Prospects. Foods 2021, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.Y.; Hou, C.J.; Bian, M.H.; Shen, C.H.; Zhang, S.Y.; Huo, D.Q.; Ma, Y. Characterization of microbial community profiles associated with quality of Chinese strong-aromatic liquor through metagenomics. J. Appl. Microbiol. 2019, 127, 750–762. [Google Scholar] [CrossRef]
- Chai, L.-J.; Xu, P.-X.; Qian, W.; Zhang, X.-J.; Ma, J.; Lu, Z.-M.; Wang, S.-T.; Shen, C.-H.; Shi, J.-S.; Xu, Z.-H. Profiling the Clostridia with butyrate-producing potential in the mud of Chinese liquor fermentation cellar. Int. J. Food Microbiol. 2019, 297, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Fan, Z.; Du, A.; Li, Y.; Zhang, R.; Shi, Q.; Shi, L.; Chu, X. Recent advances in Baijiu analysis by chromatography based technology-A review. Food Chem. 2020, 324, 126899. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, B. Effect of Fermentation Processing on the Flavor of Baijiu. J. Agric. Food Chem. 2018, 66, 5425–5432. [Google Scholar] [CrossRef]
- Xie, X.; Chen, L.; Chen, T.; Yang, F.; Wang, Z.; Hu, Y.; Lu, J.; Lu, X.; Li, Q.; Zhang, X.; et al. Profiling and annotation of carbonyl compounds in Baijiu Daqu by chlorine isotope labeling-assisted ultrahigh-performance liquid chromatography-high resolution mass spectrometry. J. Chromatogr. A 2023, 1703, 464110. [Google Scholar] [CrossRef]
- Yu, Y.; Nie, Y.; Chen, S.; Xu, Y. Characterization of the dynamic retronasal aroma perception and oral aroma release of Baijiu by progressive profiling and an intra-oral SPME combined with GC × GC-TOFMS method. Food Chem. 2023, 405, 134854. [Google Scholar] [CrossRef]
- Sharma, A.; Rai, P.K.; Prasad, S. GC–MS detection and determination of major volatile compounds in Brassica juncea L. leaves and seeds. Microchem. J. 2018, 138, 488–493. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, L.; Wang, X.; Zhu, S.; You, J.; Zhao, X.-E.; Bai, Y.; Liu, H. Integration of stable isotope labeling derivatization and magnetic dispersive solid phase extraction for measurement of neurosteroids by in vivo microdialysis and UHPLC-MS/MS. Talanta 2019, 199, 97–106. [Google Scholar] [CrossRef]
- Mu, Y.; Huang, J.; Zhou, R.; Zhang, S.; Qin, H.; Tang, H.; Pan, Q.; Tang, H. Characterization of the differences in aroma-active compounds in strong-flavor Baijiu induced by bioaugmented Daqu using metabolomics and sensomics approaches. Food Chem. 2023, 424, 136429. [Google Scholar] [CrossRef]
- Zou, W.; Zhao, C.; Luo, H. Diversity and Function of Microbial Community in Chinese Strong-Flavor Baijiu Ecosystem: A Review. Front. Microbiol. 2018, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Cao, X.; Cheng, J.; Li, L.; Zhang, T.; Wu, Q.; Xiang, P.; Shen, C.; Li, Q. Chinese Baijiu: The Perfect Works of Microorganisms. Front. Microbiol. 2022, 13, 919044. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S.; Liu, Y.; Hui, M.; Pan, C. Functional microorganisms in Baijiu Daqu: Research progress and fortification strategy for application. Front. Microbiol. 2023, 14, 1119675. [Google Scholar] [CrossRef]
- Huang, Y.; Yi, Z.; Jin, Y.; Zhao, Y.; He, K.; Liu, D.; Zhao, D.; He, H.; Luo, H.; Zhang, W.; et al. New microbial resource: Microbial diversity, function and dynamics in Chinese liquor starter. Sci. Rep. 2017, 7, 14577. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Chai, L.J.; Wang, H.M.; Lu, Z.M.; Zhang, X.J.; Xiao, C.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Bacteria and filamentous fungi running a relay race in Daqu fermentation enable macromolecular degradation and flavor substance formation. Int. J. Food Microbiol. 2023, 390, 110118. [Google Scholar] [CrossRef]
- Tan, Y.; Zhong, H.; Zhao, D.; Du, H.; Xu, Y. Succession rate of microbial community causes flavor difference in strong-aroma Baijiu making process. Int. J. Food Microbiol. 2019, 311, 108350. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, M.; Xu, B.; Liu, L.; Sun, W.; Mu, D.; Wu, X.; Li, X. Microbial communities and flavor formation in the fermentation of Chinese strong-flavor Baijiu produced from old and new Zaopei. Food Res. Int. 2022, 156, 111162. [Google Scholar] [CrossRef]
- Chai, L.J.; Qian, W.; Zhong, X.Z.; Zhang, X.J.; Lu, Z.M.; Zhang, S.Y.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Mining the Factors Driving the Evolution of the Pit Mud Microbiome under the Impact of Long-Term Production of Strong-Flavor Baijiu. Appl. Environ. Microbiol. 2021, 87, 88521. [Google Scholar] [CrossRef]
- Shoubao, Y.; Yonglei, J.; Qi, Z.; Shunchang, P.; Cuie, S. Bacterial diversity associated with volatile compound accumulation in pit mud of Chinese strong-flavor baijiu pit. AMB Express 2023, 13, 3. [Google Scholar] [CrossRef]
- Wu, L.; Fan, J.; Chen, J.; Fang, F. Chemotaxis of Clostridium Strains Isolated from Pit Mud and Its Application in Baijiu Fermentation. Foods 2022, 11, 3639. [Google Scholar] [CrossRef]
- Wang, X.J.; Zhu, H.M.; Ren, Z.Q.; Huang, Z.G.; Wei, C.H.; Deng, J. Characterization of Microbial Diversity and Community Structure in Fermentation Pit Mud of Different Ages for Production of Strong-Aroma Baijiu. Pol. J. Microbiol. 2020, 69, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.L.; Yu, Y.; Ramaswamy, H.S.; Zhu, S.M. Characterization of Chinese liquor aroma components during aging process and liquor age discrimination using gas chromatography combined with multivariable statistics. Sci. Rep. 2017, 7, 39671. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Chai, L.J.; Fang, G.Y.; Lu, Z.M.; Zhang, X.J.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Metabolite-Based Mutualistic Interaction between Two Novel Clostridial Species from Pit Mud Enhances Butyrate and Caproate Production. Appl. Environ. Microbiol. 2022, 88, 48422. [Google Scholar] [CrossRef]
- Ustick, L.J.; Larkin, A.A.; Garcia, C.A.; Garcia, N.S.; Brock, M.L.; Lee, J.A.; Wiseman, N.A.; Moore, J.K.; Martiny, A.C. Metagenomic analysis reveals global-scale patterns of ocean nutrient limitation. Science 2021, 372, 287–291. [Google Scholar] [CrossRef]
- Luo, T.; He, J.; Shi, Z.; Shi, Y.; Zhang, S.; Liu, Y.; Luo, G. Metagenomic Binning Revealed Microbial Shifts in Anaerobic Degradation of Phenol with Hydrochar and Pyrochar. Fermentation 2023, 9, 387. [Google Scholar] [CrossRef]
- Paoli, L.; Ruscheweyh, H.-J.; Forneris, C.C.; Hubrich, F.; Kautsar, S.; Bhushan, A.; Lotti, A.; Clayssen, Q.; Salazar, G.; Milanese, A.; et al. Biosynthetic potential of the global ocean microbiome. Nature 2022, 607, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Shalon, D.; Culver, R.N.; Grembi, J.A.; Folz, J.; Treit, P.V.; Shi, H.; Rosenberger, F.A.; Dethlefsen, L.; Meng, X.; Yaffe, E.; et al. Profiling the human intestinal environment under physiological conditions. Nature 2023, 617, 581–591. [Google Scholar] [CrossRef]
- Mardanov, A.V.; Gruzdev, E.V.; Beletsky, A.V.; Ivanova, E.V.; Shalamitskiy, M.Y.; Tanashchuk, T.N.; Ravin, N.V. Microbial Communities of Flor Velums and the Genetic Stability of Flor Yeasts Used for a Long Time for the Industrial Production of Sherry-like Wines. Fermentation 2023, 9, 367. [Google Scholar] [CrossRef]
- Levin, D.; Raab, N.; Pinto, Y.; Rothschild, D.; Zanir, G.; Godneva, A.; Mellul, N.; Futorian, D.; Gal, D.; Leviatan, S.; et al. Diversity and functional landscapes in the microbiota of animals in the wild. Science 2021, 372, 5352. [Google Scholar] [CrossRef]
- Carter, M.R.; Gregorich, E.G. Soil Sampling and Methods of Analysis, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 1–14. [Google Scholar] [CrossRef]
- Afiahayati; Sato, K.; Sakakibara, Y. MetaVelvet-SL: An extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning. DNA Res. 2015, 22, 69–77. [Google Scholar] [CrossRef]
- Li, S.; Chou, H.H. LUCY2: An interactive DNA sequence quality trimming and vector removal tool. Bioinformatics 2004, 20, 2865–2866. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Luo, R.; Wong, T.; Zhu, J.; Liu, C.M.; Zhu, X.; Wu, E.; Lee, L.K.; Lin, H.; Zhu, W.; Cheung, D.W.; et al. SOAP3-dp: Fast, accurate and sensitive GPU-based short read aligner. PLoS ONE 2013, 8, 65632. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Bruna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistence with Extrinsic Data. bioRxiv 2023, 1, 13. [Google Scholar] [CrossRef]
- Wei, Z.G.; Chen, X.; Zhang, X.D.; Zhang, H.; Fan, X.G.; Gao, H.Y.; Liu, F.; Qian, Y. Comparison of methods for biological sequence clustering. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 3, 13. [Google Scholar] [CrossRef]
- Abdelsalam, N.A.; Elshora, H.; El-Hadidi, M. Interactive Web-Based Services for Metagenomic Data Analysis and Comparisons. Methods Mol. Biol. 2023, 2649, 133–174. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, 587–592. [Google Scholar] [CrossRef]
- Hernández-Plaza, A.; Szklarczyk, D.; Botas, J.; Cantalapiedra, C.P.; Giner-Lamia, J.; Mende, D.R.; Kirsch, R.; Rattei, T.; Letunic, I.; Jensen, L.J.; et al. eggNOG 6.0: Enabling comparative genomics across 12,535 organisms. Nucleic Acids Res. 2023, 51, 389–394. [Google Scholar] [CrossRef]
- Gautam, A.; Zeng, W.; Huson, D.H. DIAMOND + MEGAN Microbiome Analysis. Methods Mol. Biol. 2023, 2649, 107–131. [Google Scholar] [CrossRef]
- Dong, C.; Zeng, Z.; Pu, D.K.; Wen, Q.F.; Liu, S.; Du, M.Z.; Sun, Y.; Gao, Y.Z.; Rao, N.; Huang, J.; et al. CasLocusAnno: A web-based server for annotating cas loci and their corresponding (sub)types. FEBS Lett. 2019, 593, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Hu, B.; Zhang, X.; Ge, Q.; Yan, Y.; Akresi, J.; Piyush, V.; Huang, L.; Yin, Y. dbCAN-seq update: CAZyme gene clusters and substrates in microbiomes. Nucleic Acids Res. 2023, 51, 557–563. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Classification | Total Len. (bp) | Num. | Average Len. (bp) | Max Len. (bp) | N50 Len. (bp) | ||
|---|---|---|---|---|---|---|---|
| Region | Age | Position | |||||
| Luzhou | 440 years | All-Bottom_layer | 90,762,126 | 2882 | 31,492 | 684,286 | 43,776 |
| Luzhou | 440 years | All-Middle_layer | 90,425,571 | 3338 | 27,089 | 383,109 | 34,736 |
| Luzhou | 440 years | All-Top_layer | 66,059,291 | 2301 | 28,708 | 383,189 | 36,358 |
| Luzhou | 220 years | All-Bottom_layer | 67,906,746 | 2383 | 28,496 | 938,471 | 34,156 |
| Luzhou | 220 years | All-Middle_layer | 56,765,588 | 1953 | 29,065 | 454,806 | 36,701 |
| Luzhou | 220 years | All-Top_layer | 92,198,789 | 3334 | 27,654 | 362,757 | 34,395 |
| Luzhou | 140 years | All-Bottom_layer | 65,720,000 | 2266 | 29,002 | 465,956 | 36,112 |
| Luzhou | 140 years | All-Middle_layer | 82,135,058 | 2861 | 28,708 | 544,381 | 37,429 |
| Luzhou | 140 years | All-Top_layer | 78,202,504 | 2789 | 28,039 | 353,104 | 36,019 |
| Luzhou | 50 years | All-Bottom_layer | 70,386,979 | 2204 | 31,936 | 750,639 | 43,744 |
| Luzhou | 50 years | All-Middle_layer | 65,224,763 | 2307 | 28,272 | 294,874 | 34,635 |
| Luzhou | 50 years | All-Top_layer | 82,475,229 | 2717 | 30,355 | 426,276 | 41,806 |
| Chengdu | 50 years | All-Bottom_layer | 93,242,880 | 3151 | 29,591 | 365,438 | 38,234 |
| Chengdu | 50 years | All-Middle_layer | 116,609,574 | 3931 | 29,664 | 793,643 | 38,912 |
| Chengdu | 50 years | All-Top_layer | 84,719,849 | 2791 | 30,354 | 411,342 | 40,105 |
| Mianyang | 50 years | All-Bottom_layer | 97,967,763 | 3005 | 32,601 | 879,267 | 44,720 |
| Mianyang | 50 years | All-Middle_layer | 72,020,509 | 1928 | 37,355 | 689,896 | 56,266 |
| Mianyang | 50 years | All-Top_layer | 93,155,296 | 3181 | 29,284 | 644,239 | 36,710 |
| Sample Classification | Number of Genes | Number of Unigenes | ||
|---|---|---|---|---|
| Region | Age | Position | ||
| Luzhou | 440 years | All-Bottom_layer | 86,449 | 85,431 |
| Luzhou | 440 years | All-Middle_layer | 86,338 | 85,295 |
| Luzhou | 440 years | All-Top_layer | 63,079 | 62,273 |
| Luzhou | 220 years | All-Bottom_layer | 64,754 | 63,952 |
| Luzhou | 220 years | All-Middle_layer | 54,240 | 53,577 |
| Luzhou | 220 years | All-Top_layer | 88,622 | 87,547 |
| Luzhou | 140 years | All-Bottom_layer | 62,356 | 61,522 |
| Luzhou | 140 years | All-Middle_layer | 78,894 | 77,925 |
| Luzhou | 140 years | All-Top_layer | 74,567 | 73,631 |
| Luzhou | 50 years | All-Bottom_layer | 68,442 | 67,632 |
| Luzhou | 50 years | All-Middle_layer | 62,357 | 61,567 |
| Luzhou | 50 years | All-Top_layer | 78,485 | 77,561 |
| Chengdu | 50 years | All-Bottom_layer | 88,101 | 86,959 |
| Chengdu | 50 years | All-Middle_layer | 109,015 | 107,659 |
| Chengdu | 50 years | All-Top_layer | 80,540 | 79,564 |
| Mianyang | 50 years | All-Bottom_layer | 92,053 | 90,946 |
| Mianyang | 50 years | All-Middle_layer | 67,106 | 66,184 |
| Mianyang | 50 years | All-Top_layer | 87,530 | 86,435 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.; Deng, Y.; Huang, J.; Zeng, C.; Wu, H.; Qin, H.; Zhang, S. Integrated Metagenomics and Network Analysis of Metabolic Functional Genes in the Microbial Community of Chinese Fermentation Pits. Fermentation 2023, 9, 772. https://doi.org/10.3390/fermentation9080772
Guo M, Deng Y, Huang J, Zeng C, Wu H, Qin H, Zhang S. Integrated Metagenomics and Network Analysis of Metabolic Functional Genes in the Microbial Community of Chinese Fermentation Pits. Fermentation. 2023; 9(8):772. https://doi.org/10.3390/fermentation9080772
Chicago/Turabian StyleGuo, Mingyi, Yan Deng, Junqiu Huang, Chuantao Zeng, Huachang Wu, Hui Qin, and Suyi Zhang. 2023. "Integrated Metagenomics and Network Analysis of Metabolic Functional Genes in the Microbial Community of Chinese Fermentation Pits" Fermentation 9, no. 8: 772. https://doi.org/10.3390/fermentation9080772