
Omics Sequencing of Saccharomyces cerevisiae Strain with Improved Capacity for Ethanol Production
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Detection of the Fermentation Process
2.3. Whole Genome DNA Extraction, Library Construction, and Sequencing
2.4. RNA Extraction, Library Construction, and Sequencing
2.5. Sequence Data Analysis
2.6. Quantitative Reverse Transcription PCR (qRT-PCR)
3. Results
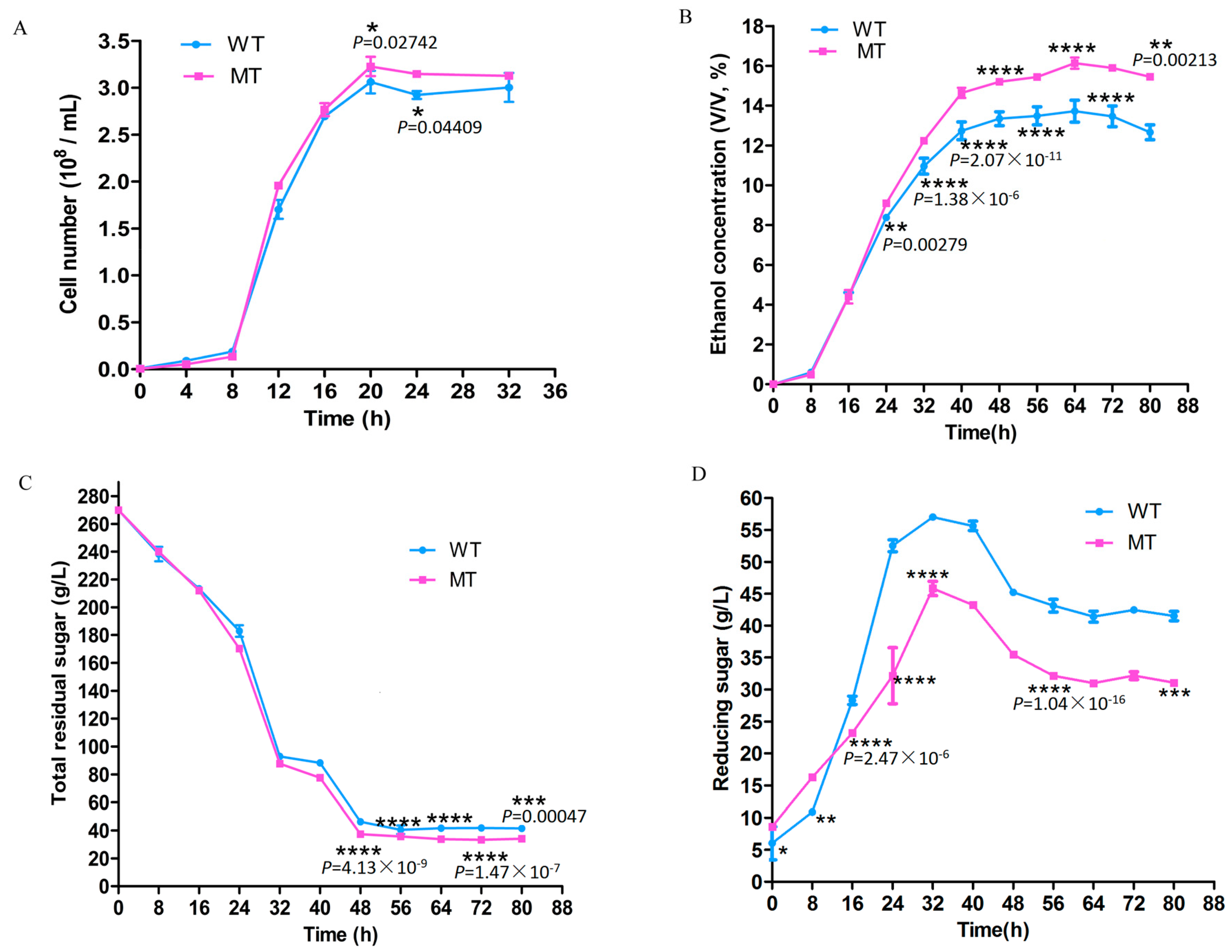
3.1. Cell Growth and Fermentation
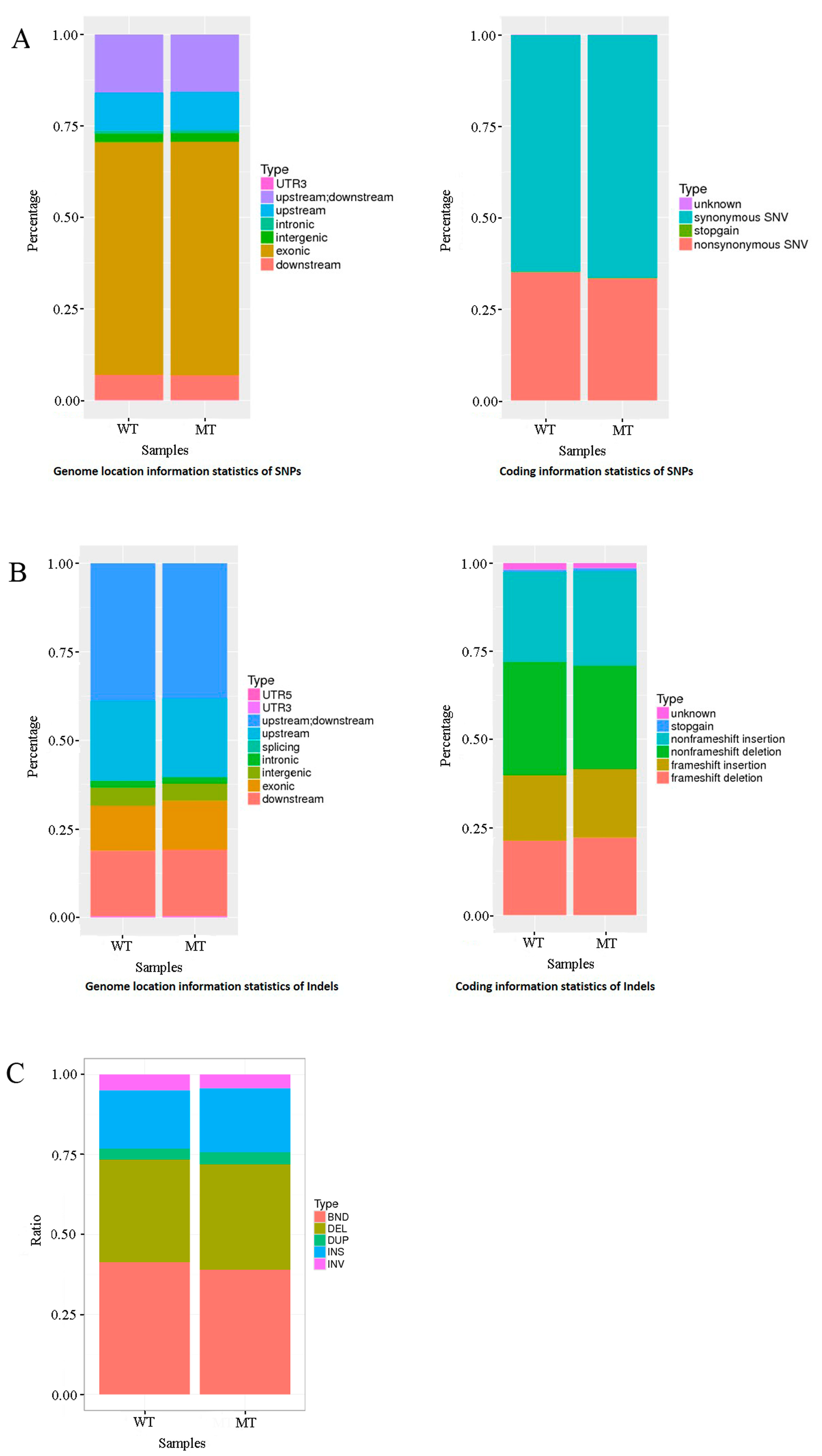
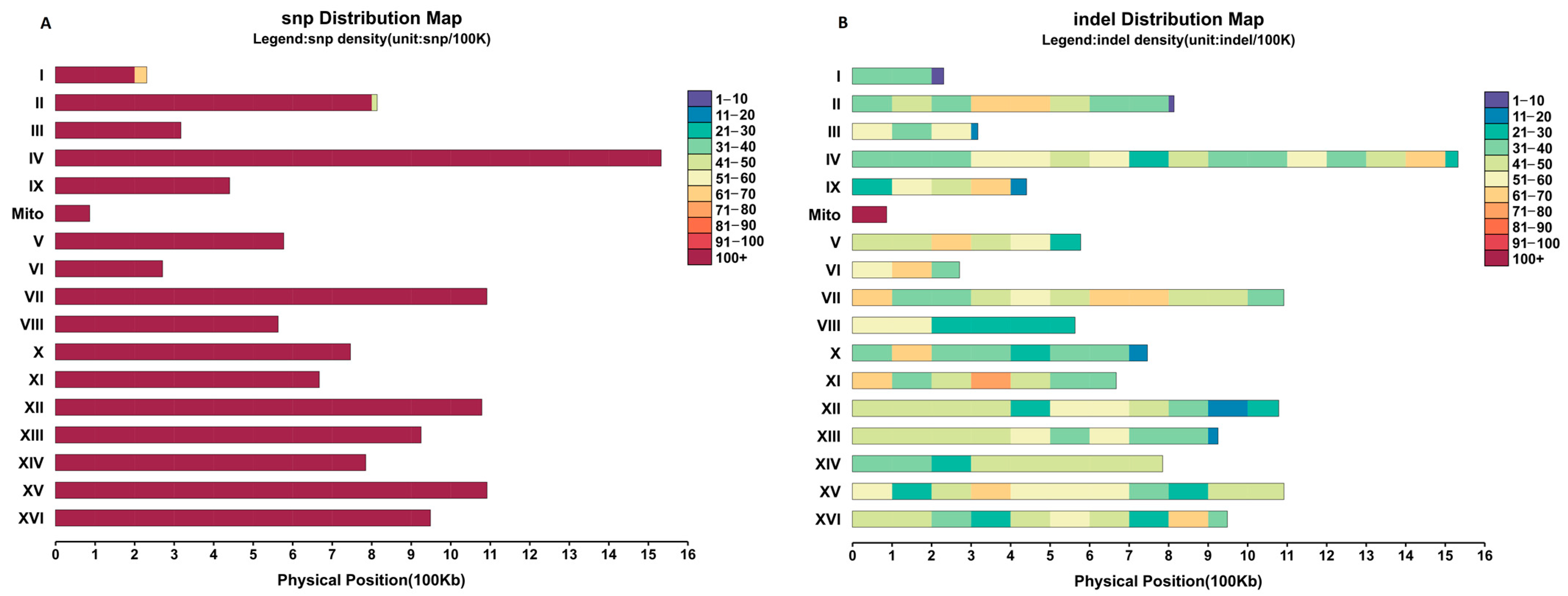
3.2. Genome Sequencing
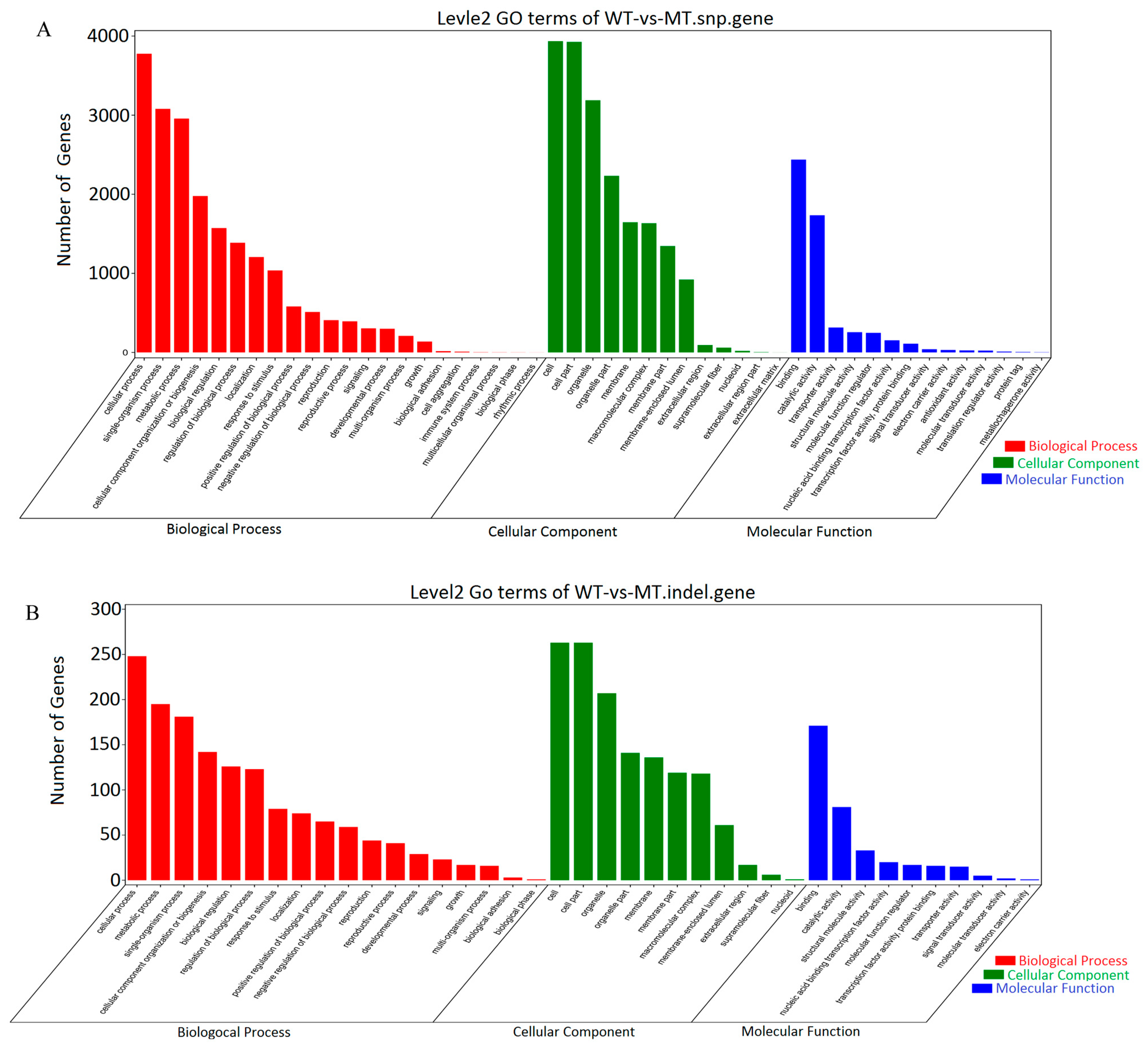
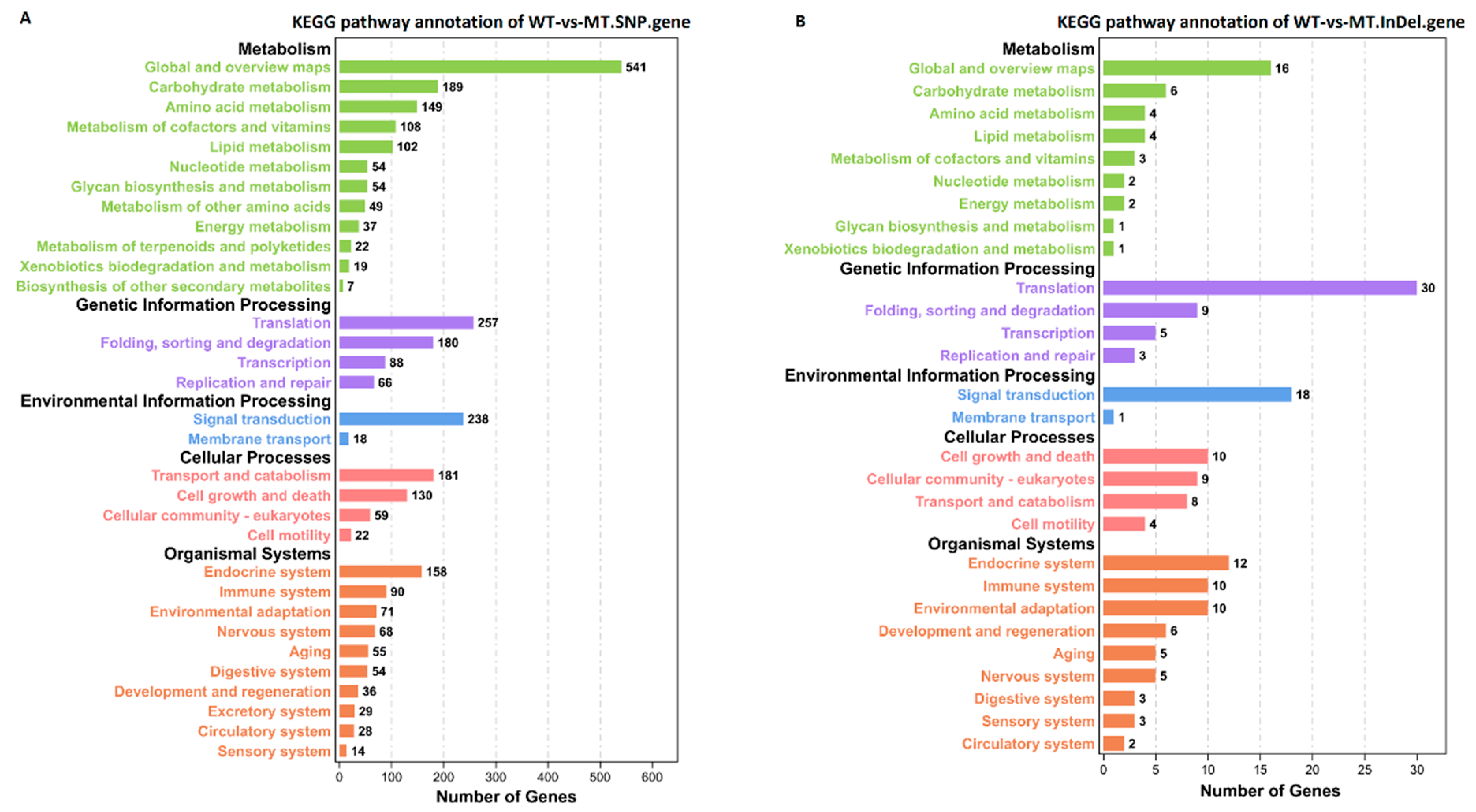
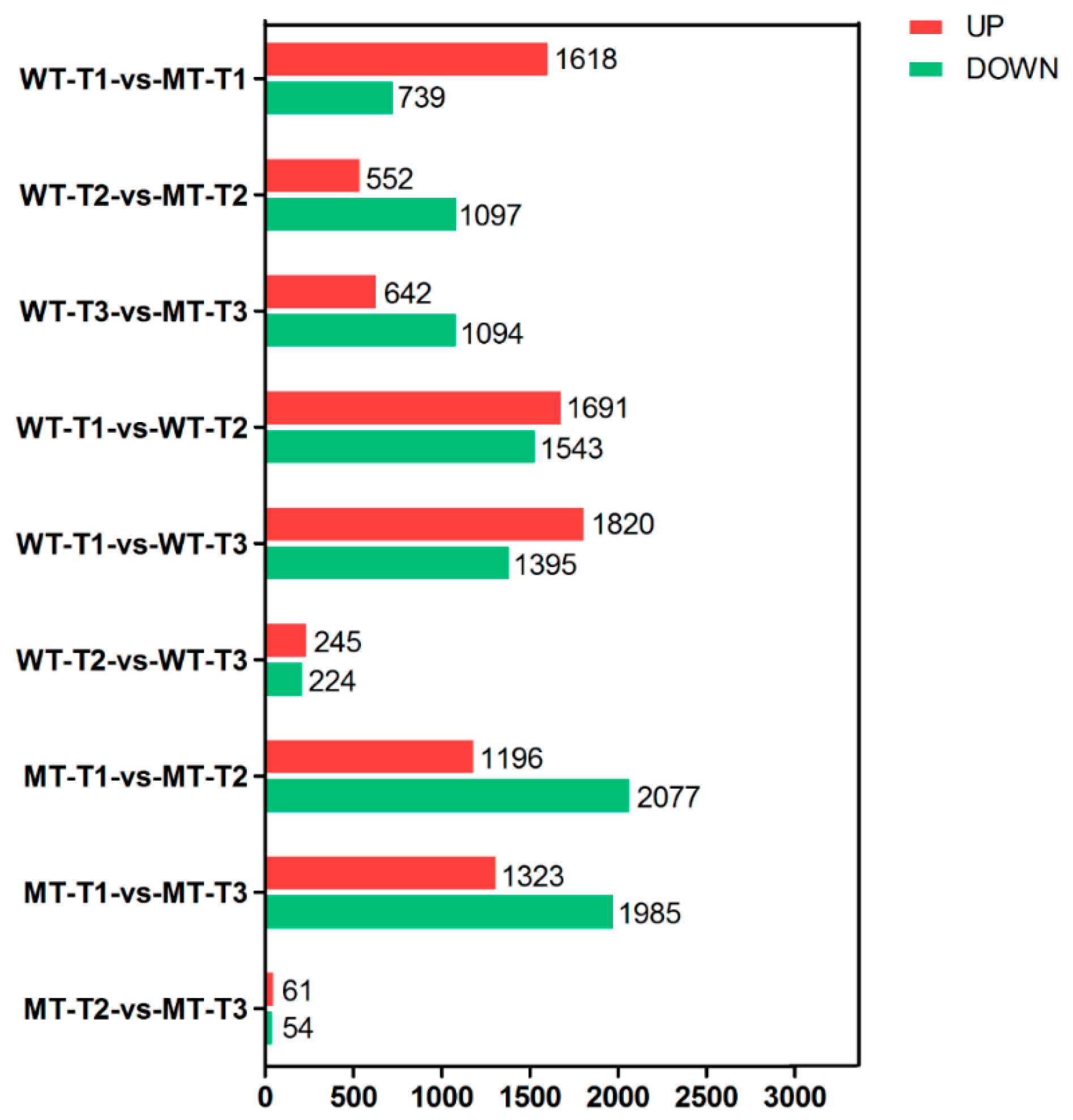
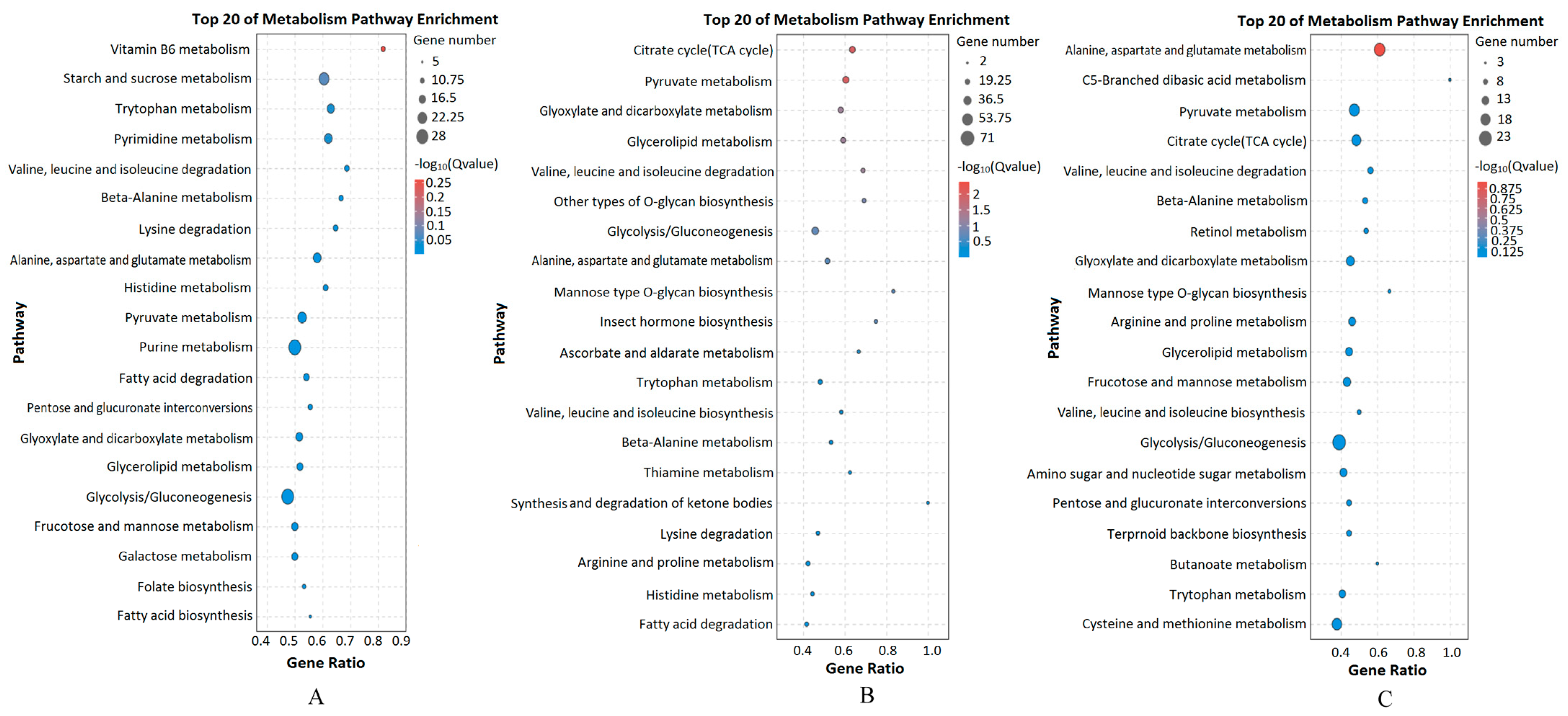
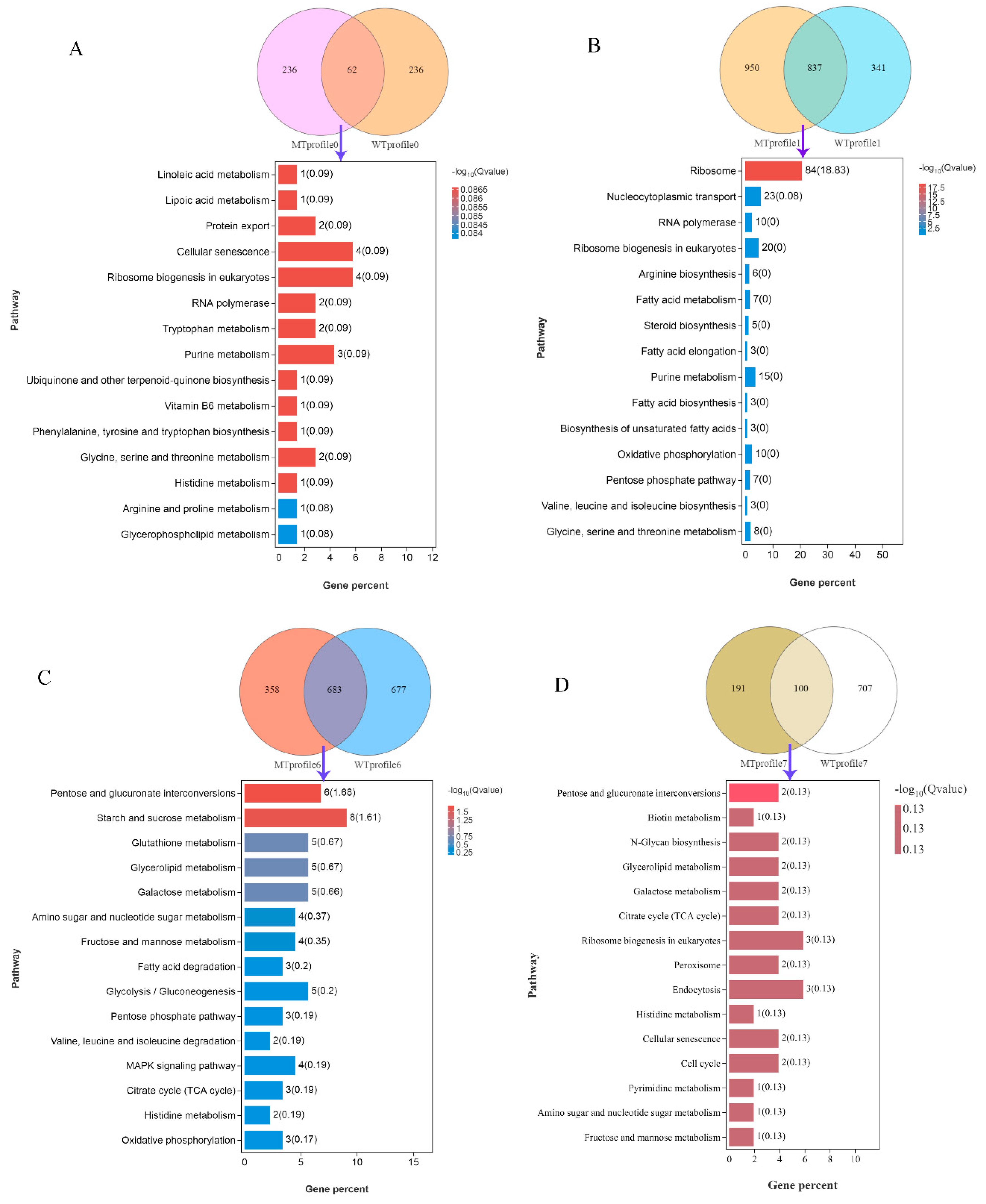
3.3. DEGs and KEGG Enrichment Analysis between Different Groups
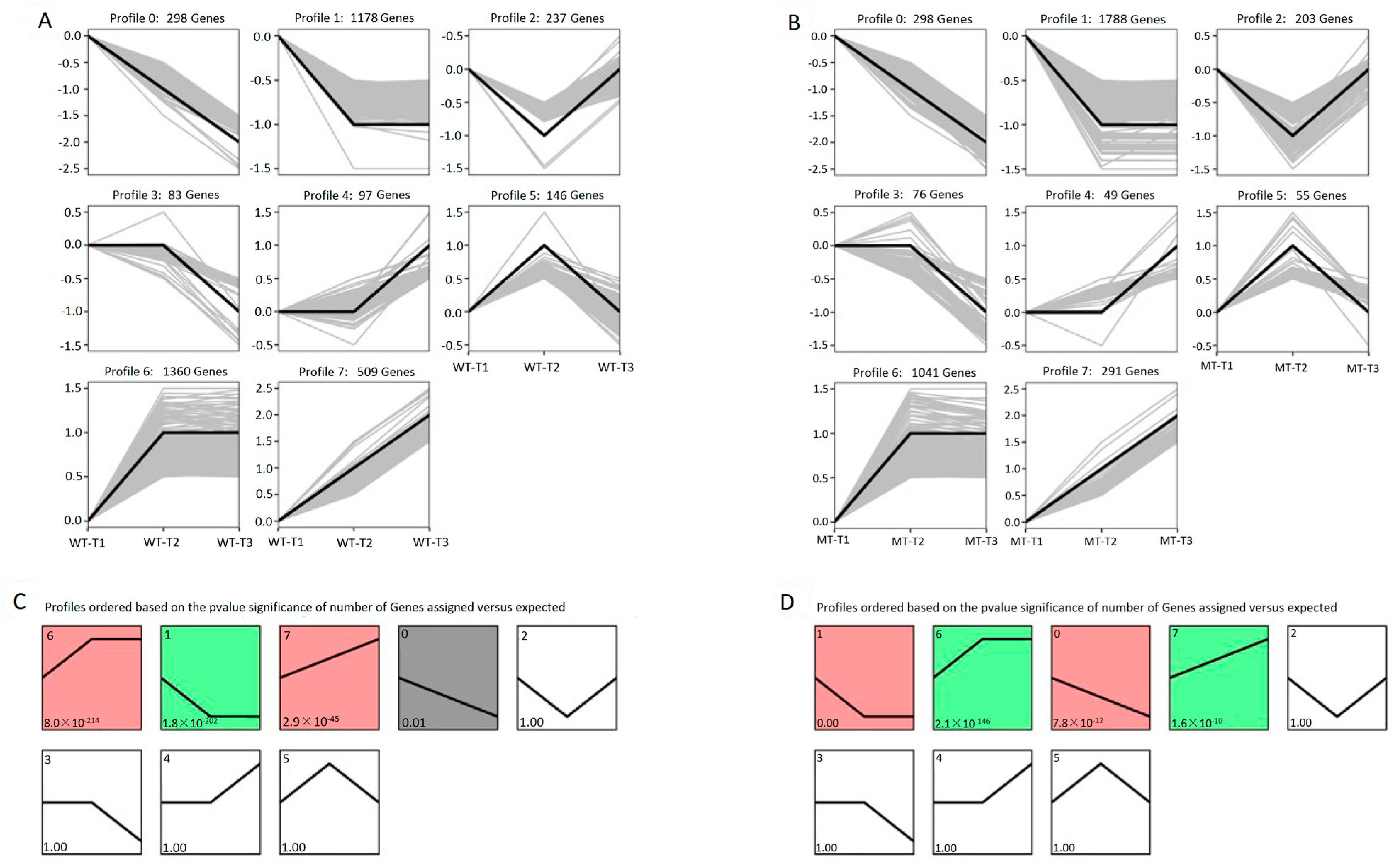
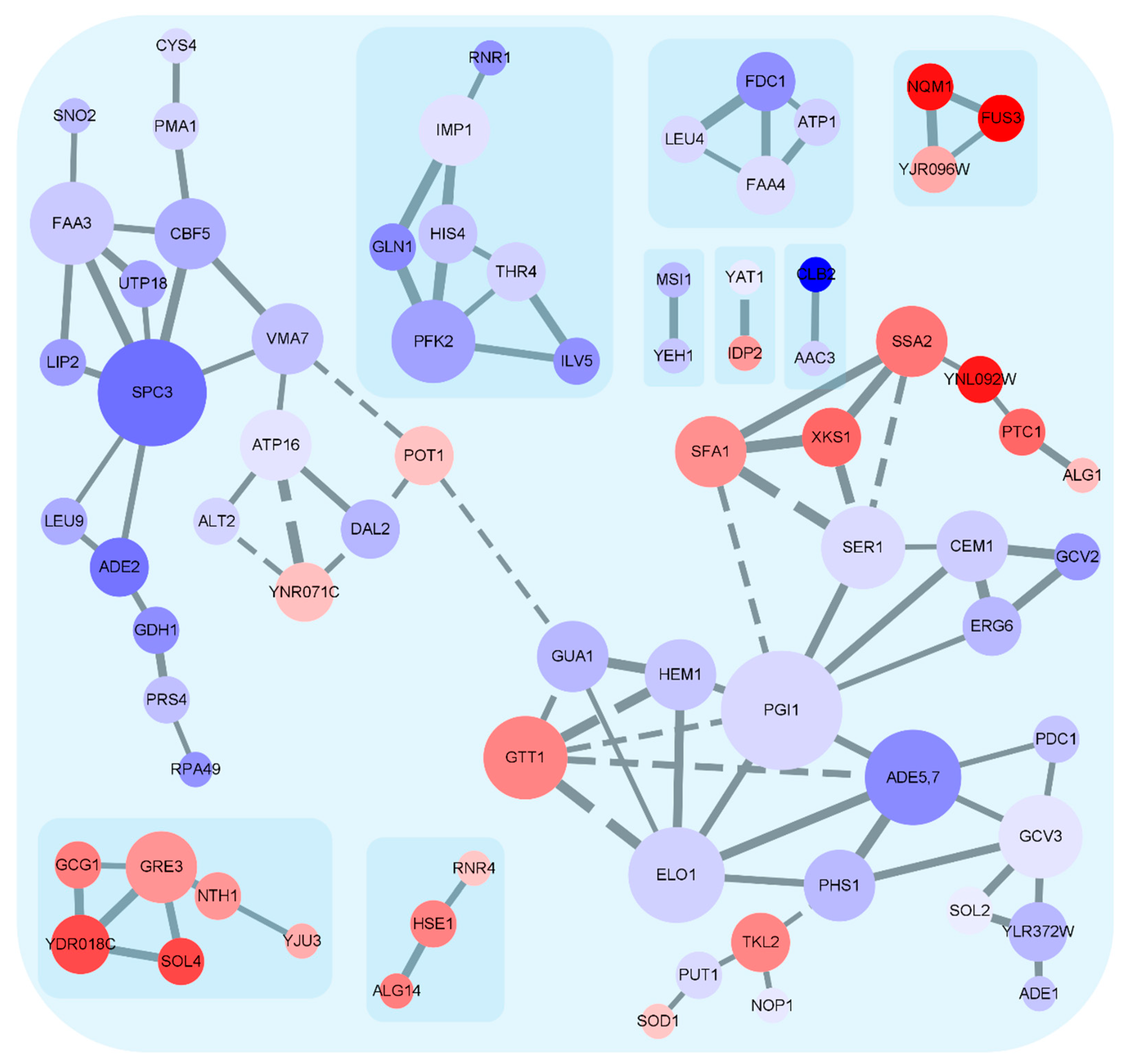
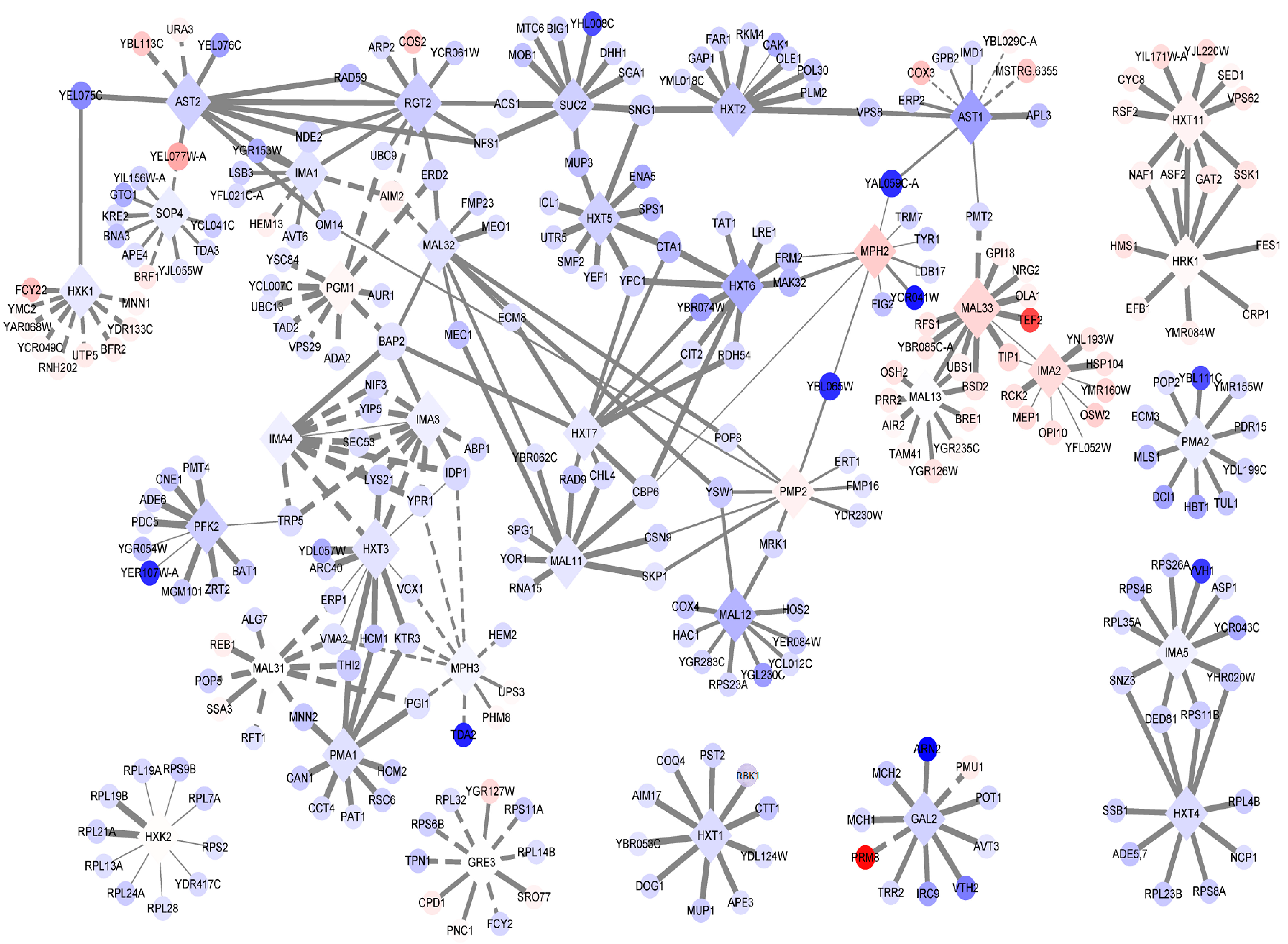
3.4. Expression Trends of DEGs on the Timeline and Exclusive Genes in the MT Strain
3.5. qRT-PCR
4. Discussion
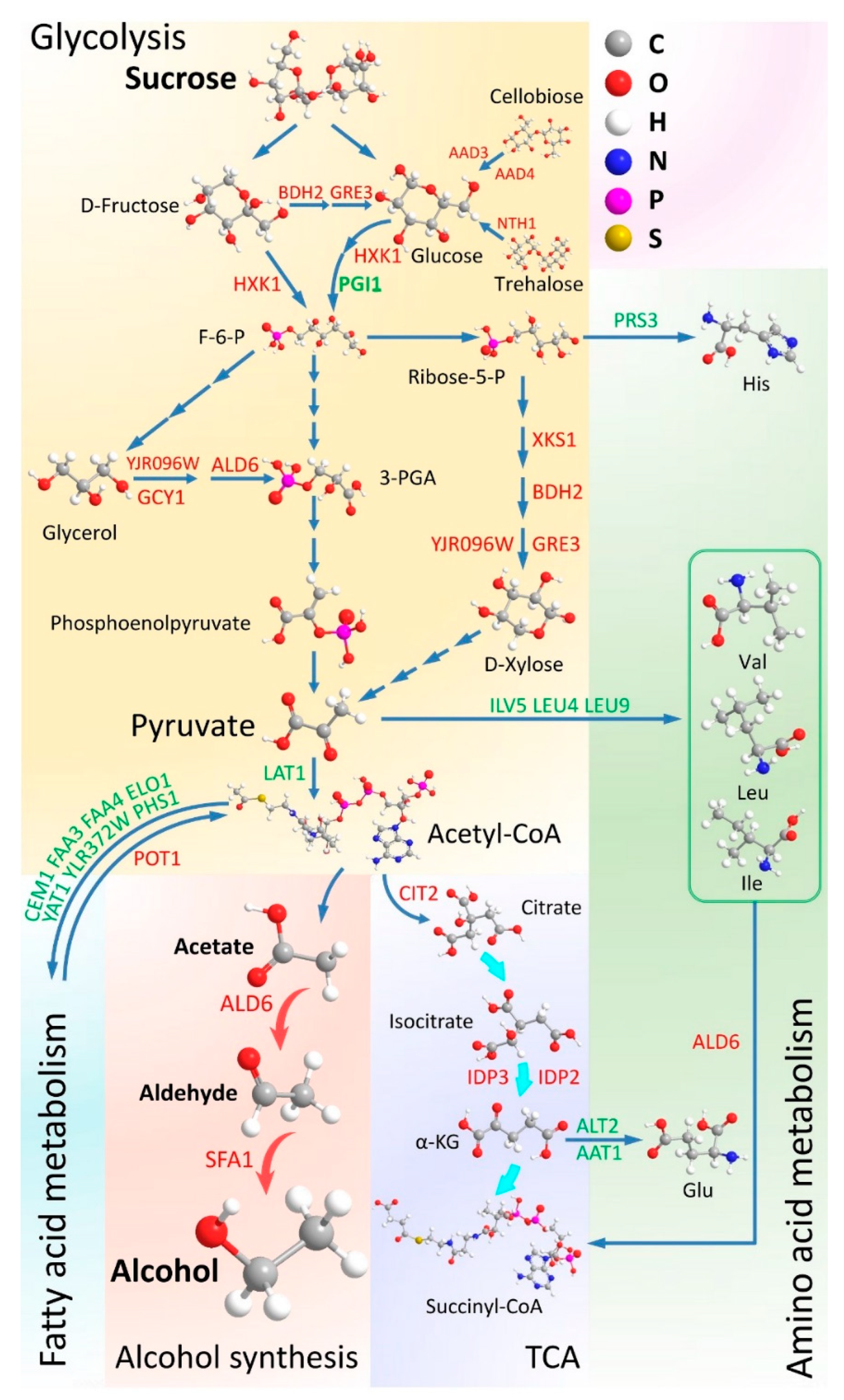
4.1. Sucrose Consumption of the WT and MT Strains
4.2. Carbon Metabolism in the MT Strain
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, X.; Zhang, S.; Guo, X.; Yang, Z.; Liu, J.; He, L.; Zheng, X.; Han, L.; Liu, H.; Wu, Y. Assessment of ethanol blended fuels for gasoline vehicles in China: Fuel economy, regulated gaseous pollutants and particulate matter. Environ. Pollut. 2019, 253, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Graeme, M.; Walker, T.O.B. Mitigating stress in industrial yeasts. Fungal Biol. 2020, 124, 387–397. [Google Scholar] [CrossRef]
- da Silva Fernandes, F.; de Souza, É.S.; Carneiro, L.M.; Alves Silva, J.P.; de Souza, J.V.B.; da Silva Batista, J. Current Ethanol Production Requirements for the Yeast Saccharomyces cerevisiae. Int. J. Microbiol. 2022, 2022, 7878830. [Google Scholar] [CrossRef]
- Gombert, A.K.; van Maris, A.J. Improving conversion yield of fermentable sugars into fuel ethanol in 1st generation yeast-based production processes. Curr. Opin. Biotechnol. 2015, 33, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Marques, W.L.; Raghavendran, V.; Stambuk, B.U.; Gombert, A.K. Sucrose and Saccharomyces cerevisiae: A relationship most sweet. FEMS Yeast Res. 2016, 16, fov107. [Google Scholar] [CrossRef]
- Della-Bianca, B.E.; Basso, T.O.; Stambuk, B.U.; Basso, L.C.; Gombert, A.K. What do we know about the yeast strains from the Brazilian fuel ethanol industry? Appl. Microbiol. Biotechnol. 2013, 97, 979–991. [Google Scholar] [CrossRef]
- Lata, K.; Sharma, M.; Patel, S.N.; Sangwan, R.S.; Singh, S.P. An integrated bio-process for production of functional biomolecules utilizing raw and by-products from dairy and sugarcane industries. Bioprocess Biosyst. Eng. 2018, 41, 1121–1131. [Google Scholar] [CrossRef]
- Walker, G.M.; Walker, R.S. Enhancing Yeast Alcoholic Fermentations. Adv. Appl. Microbiol. 2018, 105, 87–129. [Google Scholar] [CrossRef] [PubMed]
- Parapouli, M.; Vasileiadis, A.; Afendra, A.S.; Hatziloukas, E. Saccharomyces cerevisiae and its industrial applications. AIMS Microbiol. 2020, 6, 1–32. [Google Scholar] [CrossRef]
- Jacobus, A.P.; Gross, J.; Evans, J.H.; Ceccato-Antonini, S.R.; Gombert, A.K. Saccharomyces cerevisiae strains used industrially for bioethanol production. Essays Biochem. 2021, 65, 147–161. [Google Scholar] [CrossRef]
- Doğan, A.; Demirci, S.; Aytekin, A.Ö.; Şahin, F. Improvements of Tolerance to Stress Conditions by Genetic Engineering in Saccharomyces Cerevisiae during Ethanol Production. Appl. Biochem. Biotechnol. 2014, 174, 28–42. [Google Scholar] [CrossRef]
- Patra, P.; Das, M.; Kundu, P.; Ghosh, A. Recent advances in systems and synthetic biology approaches for developing novel cell-factories in non-conventional yeasts. Biotechnol. Adv. 2021, 47, 107695. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Kumar, V.; Prasad, R.; Gaur, N.A. Engineering of Saccharomyces cerevisiae as a consolidated bioprocessing host to produce cellulosic ethanol: Recent advancements and current challenges. Biotechnol. Adv. 2022, 56, 107925. [Google Scholar] [CrossRef]
- de Jong, T.V.; Moshkin, Y.M.; Guryev, V. Gene expression variability: The other dimension in transcriptome analysis. Physiol. Genom. 2019, 51, 145–158. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, J.E.; Lee, D.Y.; Park, H.; Kim, K.H.; Park, Y.C. Multi-omic characterization of laboratory-evolved Saccharomyces cerevisiae HJ7-14 with high ability of algae-based ethanol production. Appl. Microbiol. Biotechnol. 2018, 102, 8989–9002. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Hsu, T.; Sirota-Madi, A.; Shafquat, A.; Abu-Ali, G.; Morgan, X.C.; Huttenhower, C. Sequencing and beyond: Integrating molecular 'omics' for microbial community profiling. Nat. Rev. Microbiol. 2015, 13, 360–372. [Google Scholar] [CrossRef]
- Peng, D.; Ruan, C.; Fu, S.; He, C.; Song, J.; Li, H.; Tu, Y.; Tang, D.; Yao, L.; Lin, S.; et al. Atg9-centered multi-omics integration reveals new autophagy regulators in Saccharomyces cerevisiae. Autophagy 2021, 17, 4453–4476. [Google Scholar] [CrossRef]
- Yang, Y.; Ren, W.; Xu, H.; Cheng, L.; Dapaah, M.F.; He, R.; Ma, H. Incorporating Transcriptomic-Metabolomic analysis reveal the effect of ultrasound on ethanol production in Saccharomyces Cerevisiae. Ultrason. Sonochem. 2021, 79, 105791. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Wu, Y.; Chen, Y.; Chen, X.; Wu, R.; Lu, Q.; Chen, D.; Huang, R. Role of spt23 in Saccharomyces cerevisiae thermal tolerance. Appl. Microbiol. Biotechnol. 2022, 106, 3691–3705. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Litvinchuk, A.V.; Sokolov, S.S.; Rogov, A.G.; Markova, O.V.; Knorre, D.A.; Severin, F.F. Mitochondrially-encoded protein Var1 promoter loss of respiratory function in Saccharomyces cerevisiae under stressful conditions. Eur. J. Cell Biol. 2013, 92, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.J.; Kim, J.E.H.; Junne, T.; Spiess, M.; Kim, H. Cotranslational Targeting and Posttranslational Translocation can Cooperate in Spc3 Topogenesis. J. Mol. Biol. 2021, 433, 167109. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Maity, S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk—Signaling Beyond (ER) Stress Response. Biomolecules 2021, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.R.; Staff, M.; Lee, R.; Love, J.; Parker, D.A.; Aves, S.J.; Howard, T.P. Design of experiments methodology to build a multifactorial statistical model describing the metabolic interactions of alcohol. ACS Synth. Biol. 2018, 7, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Mariani, D.; Mathias, C.J.; da Silva, C.G.; Herdeiro, R.D.S.; Pereira, R.; Panek, A.D.; Eleutherio, E.C.; Pereira, M.D. Involvement of glutathione transferases, Gtt1and Gtt2, with oxidative stress response generated by H2O2 during growth of Saccharomyces cerevisiae. Redox Rep. 2008, 13, 246–254. [Google Scholar] [CrossRef]
- Raghavendran, V.; Marx, C.; Olsson, L.; Bettiga, M. The protective role of intracellular glutathione in Saccharomyces cerevisiae during lignocellulosic ethanol production. AMB Express 2020, 10, 219. [Google Scholar] [CrossRef]
- Kim, S.R.; Kwee, N.R.; Kim, H.; Jin, Y.S. Feasibility of xylose fermentation by engineered Saccharomyces cerevisiae overexpressing endogenous aldose reductase (GRE3), xylitol dehydrogenase (XYL2), and xylulokinase (XYL3) from Scheffersomyces stipitis. FEMS Yeast Res. 2013, 13, 312–321. [Google Scholar] [CrossRef]
- Marques, W.L.; Mans, R.; Marella, E.R.; Cordeiro, R.L.; Broek, M.; Daran, J.M.G.; Pronk, J.T.; Gombert, A.K.; van Maris, A.J. Elimination of Elimination of sucrose transport and hydrolysis in Saccharomyces cerevisiae: A platform strain for engineering sucrose metabolism. FEMS Yeast Res. 2017, 17, fox006. [Google Scholar] [CrossRef]
- Carlson, M.; Botstein, D. Two differentially regulated mRNAs with different 5' ends encode secreted with intracellular forms of yeast invertase. Cell 1982, 28, 145–154. [Google Scholar] [CrossRef]
- Perlman, D.; Halvorson, H.O.; Cannon, L.E. Presecretory and cytoplasmic invertase polypeptides encoded by distinct mRNAs derived from the same structural gene differ by a signal sequence. Proc. Natl. Acad. Sci. USA 1982, 79, 781–785. [Google Scholar] [CrossRef]
- de Valk, S.C.; Bouwmeester, S.E.; de Hulster, E.; Mans, R. Engineering proton-coupled hexose uptake in Saccharomyces cerevisiae for improved ethanol yield. Biotechnol. Biofules Bioprod. 2022, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Rodriguez, R. Glucose regulation of the paralogous glucose sensing receptors Rgt2 and Snf3 of the yeast Saccharomyces cerevisiae. Biochem. Biophys. Acta. Gen. Subj. 2021, 1865, 129881. [Google Scholar] [CrossRef]
- Scharff-Poulsen, P.; Moriya, H.; Johnston, M. Genetic Analysis of Signal Generation by the Rgt2 Glucose Sensor of Saccharomyces cerevisiae. Genes Genomes Genet. 2018, 8, 2685–2696. [Google Scholar] [CrossRef]
- Diniz, A.L.; da Silva, D.I.R.; Lembke, C.G.; Costa, M.D.B.L.; Ten-Caten, F.; Li, F.; Vilela, R.D.; Menossi, M.; Ware, D.; Endres, L.; et al. Amino Acid and Carbohydrate Metabolism Are Coordinated to Maintain Energetic Balance during Drought in Sugarcane. Int. J. Mol. Sci. 2020, 21, 9124. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Alam, M.T.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2014, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Khan, A.; Shen, Y.; Chen, L.; Rabinowitz, J.D. Glucose feeds the tricarboxylic acid cycle via excreted ethanol in fermenting yeast. Nat. Chem. Biol. 2022, 18, 1380–1387. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Gene ID | Gene Symbol |
|---|---|---|
| Profile 0 a | ||
| Linoleic acid metabolism | YOL011W | PLB3 |
| Lipoic acid metabolism | YLR239C | LIP2 |
| Protein export | YMR150C, YLR066W | IMP1, SPC3 |
| Cellular senescence | YPR119W, YBR085, WYKL168C, YBR195C | CLB2, AAC3, KKQ8, MSI1 |
| Ribosome biogenesis in eukaryotes | YLR129W, YLR175W, YDL014W, YJL069C | DIP2, CBF5, NOP1, UTP18 |
| RNA polymerase | YNL248C, YGL070C | RPA49, RPB9 |
| Tryptophan metabolism | YLR044C, YGR088W | PDC1, CTT1 |
| Purine metabolism | YEL042W, YIR029W, YOR128C | GDA1, DAL2, ADE2 |
| Ubiquinone and other terpenoid-quinone biosynthesis | YDR539W | FDC1 |
| Vitamin B6 metabolism | YNL334C | SNO2 |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | YDR007W | TRP1 |
| Glycine, serine, and threonine metabolism | YML082W, YMR189W | YML082W, GCV2 |
| Histidine metabolism | YCL030C | HIS4 |
| Arginine and proline metabolism | YLR142W | PUT1 |
| Glycerophospholipid metabolism | YOL011W | PLB3 |
| Profile 1 b | ||
| Ribosome | YGR085C, YDR418W, YIL133C, YNL069C, YDL082W, YHL001W, YJL177W, YMR242C, YOR312C, YNL301C, YBL027W, YBR191W, YFL034C-A, YOL127W, YBL087C, YER117W, YGL031C, YLR009W, YGR034W, YLR344W, YDR471W, YHR010W, YGL030W, YDL075W, YLR406C, YBL092W, YER056C-A, YIL052C, YOR234C, YPL143W, YDL191W, YDL136W, YMR194W, YPR043W, YLR185W, YDR500C, YLR325C, YKR094C, YIL148W, YNL162W, YHR141C, YML073C, YLR448C, YPL198W, YFR031C-A, YIL018W, YGL147C, YLR340W, YDL081C, YOL039W, YOR293W, YDR025W, YCR031C, YDR337W, YLR367W, YOL040C, YDL083C, YMR143W, YDR447C, YML024W, YML026C, YDR450W, YNL302C, YOL121C, YJL136C, YKR057W, YPR132W, YER074W, YLR333C, YGR027C, YHR021C, YLR264W, YOR167C, YDL061C, YOR182C, YNL178W, YHR203C, YPL090C, YBR181C, YNL096C, YBL072C, YBR189W, YGR214W, YLR048W | RPL11B, RPL12B, RPL16A, RPL16B, RPL13A, RPL14B, RPL17B, RPL20A, RPL20B, RPL18B, RPL19B, RPL21A, RPL22B, RPL25, RPL23A, RPL23B, RPL24A, RLP24, RPL26B, RPL26A, RPL27B, RPL27A, RPL30, RPL31A, RPL31B, RPL32, RPL34A, RPL34B, RPL33B, RPL33A, RPL35A, RPL35B, RPL36A, RPL43A, RPL37A, RPL37B, RPL38, RPL40B, RPL40A, RPL42A, RPL42B, RPL6A, RPL6B, RPL7B, RPL2A, RPL2B, RPL9A, RPP0, RPP1A, RPP2A, RPS10A, RPS11A, RPS14A, MRPS28, RPS22B, RPS15, RPS16B, RPS16A, RPS17B, RPS17A, RPS18B, RPS18A, RPS19B, RPS19A, RPS21B, RPS21A, RPS23B, RPS24A, RPS25B, RPS25A, RPS27B, RPS28B, RPS28A, RPS29B, RPS30B, RPS3, RPS4B, RPS6A, RPS6B, RPS7B, RPS8A, RPS9B, RPS0A, RPS0B |
| Nucleocytoplasmic transport | YER168C, YNL221C, YNL244C, YMR260C, YJR007W, YOR260W, YER025W, YMR146C, TDR429C, YMR309C, YOR361C, YBR079C, YJL138C, YOL139C, YGR162W, YPR041W, YGR083C, YLR208W, YKL205W, YGL092W, YKL068W, YFR002W, YMR235C | CCA1, POP1, SUI1, TIF11, SUI2, GCD1, GCD11, TIF34, TIF35, NIP1, PRT1, RPG1, TIF2, CDC33, TIF4631, TIF5, GCD2, SEC13, LOS1, NUP145, NUP100, NIC96, RNA1 |
| RNA polymerase | YOR341W, YJR063W, YPR010C, YIL021W, YPR187W, YOR224C, YNL113W, YNR003C, YDL150W, YPR110C | RPA190, RPA12, RPA135, RPB3, RPO26, RPB8, RPC19, RPC34, RPC53, RPC40 |
| Ribosome biogenesis in eukaryotes | YNL221C, YIL035C, YHR089C, YDL208W, YLR059C, YNL132W, YNL163C, YGR090W, YCL031C, YDR398W, YDR324C, YMR093W, YLR222C, YCR057C, YLR197W, YDR339C, YML093W, YLR186W, YLR022C, YLR397C | POP1, CKA1, GAR1, NHP2, REX2, KRE33, RIA1, UTP22, RRP7, UTP5, UTP4, UTP15, UTP13, PWP2, NOP56, FCF1, UTP14, EMG1, SDO1, AFG2 |
| Arginine biosynthesis | YOR375C, YDR111C, YPR035W, YOL058W, YER069W, YKL106W | ARG5,6, ALT2, GDH1, GLN1, ARG1, AAT1 |
| Fatty acid metabolism | YIL009W, YMR246W, YAR035W, YER061C, YLR372W, YJL196C, YJL097W | FAA3, FAA4, YAT1, CEM1, YLR372W, ELO1, PHS1 |
| Steroid biosynthesis | YLR056W, YLR020C, YML008C, YLL012W, YGR060W | ERG3, YEH2, ERG6, YEH1, ERG25 |
| Fatty acid elongation | YLR372W, YJL196C, YJL097W | YLR372W, ELO1, PHS1 |
| Purine metabolism | YJR105W, YJR069C, YBL068W, YMR120C, YER070W, YMR300C, YJL070C, YML022W, YMR217W, YER005W, AR015W, YGL234W, YHL011C, YCR026C, YNL141W | ADO1, HAM1, PRS4, ADE17, RNR1, ADE4, YMR120C, APT1, GUA1, YND1, ADE1, ADE5,7, PRS3, NPP1, AAH1 |
| Fatty acid biosynthesis | YIL009W, YMR246W, YER061C | FAA3, FAA4, CEM1 |
| Biosynthesis of unsaturated fatty acids | YLR372W, YJL196C, YJL097W | YLR372W, ELO1, PHS1 |
| Oxidative phosphorylation | YOR270C, YBR039W, YDL004W, YGL008C, YKL141W, YGR020C, YLR447C, YDL185W, YBL099W, YDR529C | VPH1, ATP3, ATP16, PMA1, SDH3, VMA7, VMA1, ATP1, QCR7 |
| Pentose phosphate pathway | YBL068W, YMR205C, YCR073W-A, YGR240C, YGL185C, YBR196C, YHL011C | PRS4, PFK2, SOL2, PFK1, YGL185C, PGI1, PRS3 |
| Valine, leucine, and isoleucine biosynthesis | YNL104C, YOR108W, YLR355C | LEU4, LEU9, ILV5 |
| Glycine, serine, and threonine metabolism | YDR232W, YDR158W, YOL056W, YCR053W, YOR184W, YGL185C, YGR155W, YAL044C | HEM1, HOM2, GPM3, THR4, SER1, YGL185C, CYS4, GCV3 |
| Profile 6 c | ||
| Pentose and glucuronate interconversions | YGR194C, YKL035W, YJR096W, YAL061W, YHL012W, YHR104W | XKS1, UGP1, YJR096W, BDH2, YHL012W, GRE3 |
| Starch and sucrose metabolism | YDL243C, YCR107W, YKL035W, YEL011W, YDR001C, YFR053C, YHL012W, YLR258W | AAD4, AAD3, UGP1, GLC3, NTH1, HXK1, YHL012W, GSY2 |
| Glutathione metabolism | YER163C, YGR180C, YLR174W, YIR038C, YKL026C | GCG1, RNR4, IDP2, GTT1, GPX1 |
| Glycerolipid metabolism | YDR018C, YKL094W, YPL061W, YJR096W, YHR104W | YDR018C, YJU3, ALD6, YJR096W, GRE3 |
| Galactose metabolism | YKL035W, YJR096W, YFR053C, YHL012W, YHR104W | UGP1, YJR096W, HXK1, YHL012W, GRE3 |
| Amino sugar and nucleotide sugar metabolism | YKL035W, YMR084W, YFR053C, YHL012W | UGP1, YMR084W, HXK1, YHL012W |
| Fructose and mannose metabolism | YJR096W, YFR053C, YAL061W, YHR104W | YJR096W, HXK1, BDH2, GRE3 |
| Fatty acid degradation | YPL061W, YDL168W, YIL160C | ALD6, SFA1, POT1 |
| Glycolysis/Gluconeogenesis | YDL243C, YCR107W, YPL061W, YDL168W, YFR053C | AAD4, AAD3, ALD6, SFA1, HXK1 |
| Pentose phosphate pathway | YGR248W, YBR117C, YGR043C | SOL4, TKL2, NQM1 |
| Valine, leucine and isoleucine degradation | YPL061W, YIL160C | ALD6, POT1 |
| MAPK signaling pathway | YDL006W, YNL098C, YBL016W, YLL024C | PTC1, RAS2, FUS3, SSA2 |
| Citrate cycle (TCA cycle) | YLR164W, YLR174W, YJL045W | SHH4, IDP2, YJL045W |
| Histidine metabolism | YPL061W, YNL092W | ALD6, YNL092W |
| Oxidative phosphorylation | YLR164W, Q0130, YJL045W | SHH4, OLI1, YJL045W |
| Profile 7 d | ||
| Pentose and glucuronate interconversions | YOR120W, YNR073C | GCY1, MAN2 |
| Biotin metabolism | YNR058W | BIO3 |
| N-Glycan biosynthesis | YBR110W, YBR070C | ALG1, ALG14 |
| Glycerolipid metabolism | YOR120W, YOR245C | GCY1, DGA1 |
| Galactose metabolism | YOR120W, YNR071C | GCY1, YNR071C |
| Citrate cycle (TCA cycle) | YCR005C, YNL009W | CIT2, IDP3 |
| Ribosome biogenesis in eukaryotes | YHR170W, YNL075W, YLR106C | NMD3, IMP4, MDN1 |
| Peroxisome | YNL009W, YJR104C | IDP3, SOD1 |
| Endocytosis | YHL002W, YOR211C, YER125W | HSE1, MGM1, RSP5 |
| Histidine metabolism | YMR209C | YMR209C |
| Cellular senescence | YCR008W, YJR066W | SAT4, TOR1 |
| Cell cycle | YJL187C, YCR008W | SWE1, SAT4 |
| Pyrimidine metabolism | YDR020C | DAS2 |
| Amino sugar and nucleotide sugar metabolism | YBR023C | CHS3 |
| Fructose and mannose metabolism | YOR120W | GCY1 |
| Gene Name | Description | FDR | Log2-Fold Change a |
|---|---|---|---|
| SUC2 | Invertase; sucrose hydrolyzing enzyme | 1.08 × 10−20 | −2.28 |
| MAL11 | High-affinity maltose transporter (alpha-glucoside transporter) | 0.205 | −0.91 |
| MAL31 | Maltose permease; high-affinity maltose transporter (alpha-glucoside transporter) | 6.65 × 10−5 | 0.72 |
| MAL12 | Maltase (alpha-D-glucosidase) | 0.141 | −4.03 |
| MAL32 | Maltase (alpha-D-glucosidase) | 0.466 | −1.29 |
| MAL33 | MAL-activator protein | 5.67 × 10−28 | 2.87 |
| MAL13 | MAL-activator protein | 6.15 × 10−4 | 0.55 |
| PMA1 | Plasma membrane P2-type H+-ATPase | 1.98 × 10−4 | −1.26 |
| PMA2 | Plasma membrane H+-ATPase | 0.925 | −0.53 |
| PMP2 | Proteolipid associated with plasma membrane H(+)-ATPase (Pma1p) | 0.379 | 1.39 |
| SOP4 | ER-membrane protein | 0.935 | −0.46 |
| AST1 | Lipid raft-associated protein; interacts with the plasma membrane ATPase Pma1p and has a role in its targeting of the plasma membrane by influencing its incorporation into lipid rafts | 2.32 × 10−27 | −5.37 |
| AST2 | Lipid raft-associated protein; overexpression restores Pma1p localization to lipid rafts which are required for targeting Pma1p to the plasma membrane | 1.54 × 10−5 | −2.36 |
| HRK1 | Protein kinase; implicated in activation of the plasma membrane H(+)-ATPase Pma1p in response to glucose metabolism | 1.4 × 10−8 | 1.00 |
| IMA1 | Major isomaltase (alpha-1,6-glucosidase/alpha-methylglucosidase) | 0.186 | −1.25 |
| IMA2 | Isomaltase (alpha-1,6-glucosidase/alpha-methylglucosidase) | 1.28 × 10−34 | 2.58 |
| IMA3 | Alpha-glucosidase; weak but broad substrate specificity for alpha-1,4- and alpha-1,6-glucosides | 0.697 | −0.27 |
| IMA4 | Alpha-glucosidase; weak but broad substrate specificity for alpha-1,4- and alpha-1,6-glucosides | 0.697 | −0.27 |
| IMA5 | Alpha-glucosidase; specificity for isomaltose, maltose, and palatinose, but not alpha-methylglucoside | 0.813 | −0.21 |
| MPH2 | Alpha-glucoside permease | 1 | 3.32 |
| MPH3 | Alpha-glucoside permease | 0.397 | 0.087 |
| PFK2 | Beta subunit of heterooctameric phosphofructokinase | 6.89 × 10−22 | −2.41 |
| GRE3 | Aldose reductase; involved in methylglyoxal, d-xylose, arabinose, and galactose metabolism | 1.45 × 10−9 | 0.62 |
| PGM1 | Phosphoglucomutase, minor isoform; catalyzes the conversion from glucose-1-phosphate to glucose-6-phosphate | 3.2 × 10−8 | 1.16 |
| GAL2 | Galactose permease | 0.017 | −2.12 |
| RGT2 | Plasma membrane high glucose sensor that regulates glucose transport | 5.51 × 10−11 | −2.54 |
| HXT1 | Low-affinity glucose transporter of the major facilitator superfamily | 1.08 × 10−5 | −1.53 |
| HXT2 | High-affinity glucose transporter of the major facilitator superfamily | 2.19 × 10−7 | −2.84 |
| HXT3 | Low-affinity glucose transporter of the major facilitator superfamily | 0.018 | −1.08 |
| HXT4 | High-affinity glucose transporter; member of the major facilitator superfamily | 2.79 × 10−4 | −1.57 |
| HXT5 | Hexose transporter with moderate affinity for glucose | 3.54 × 10−15 | −2.31 |
| HXT6 | High-affinity glucose transporter; member of the major facilitator superfamily | 1.99 × 10−26 | −4.56 |
| HXT7 | High-affinity glucose transporter; member of the major facilitator superfamily | 0.414 | −0.83 |
| HXT11 | Hexose transporter | 2.96 × 10−11 | 1.29 |
| HXK1 | Hexokinase isoenzyme 1 | 0.228 | −0.77 |
| HXK2 | Hexokinase isoenzyme 2 | 1.30 × 10−2 | 0.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Z.; Guo, L.; Chen, X.; Lu, Q.; Wu, Y.; Chen, D.; Wu, R.; Chen, Y. Omics Sequencing of Saccharomyces cerevisiae Strain with Improved Capacity for Ethanol Production. Fermentation 2023, 9, 483. https://doi.org/10.3390/fermentation9050483
Lu Z, Guo L, Chen X, Lu Q, Wu Y, Chen D, Wu R, Chen Y. Omics Sequencing of Saccharomyces cerevisiae Strain with Improved Capacity for Ethanol Production. Fermentation. 2023; 9(5):483. https://doi.org/10.3390/fermentation9050483
Chicago/Turabian StyleLu, Zhilong, Ling Guo, Xiaoling Chen, Qi Lu, Yanling Wu, Dong Chen, Renzhi Wu, and Ying Chen. 2023. "Omics Sequencing of Saccharomyces cerevisiae Strain with Improved Capacity for Ethanol Production" Fermentation 9, no. 5: 483. https://doi.org/10.3390/fermentation9050483