
Analysis of the Microbial Diversity and Population Dynamics during the Pulque Fermentation Process

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Pulque Fermentation in the Laboratory
2.3. Physicochemical Parameters Analysis (pH, Temperature, CO2, Dissolved Oxygen), Ashes, and Biomass
2.4. HPLC Quantification: Sucrose, Glucose, Fructose, Glycerol, Organic Acids, and Ethanol
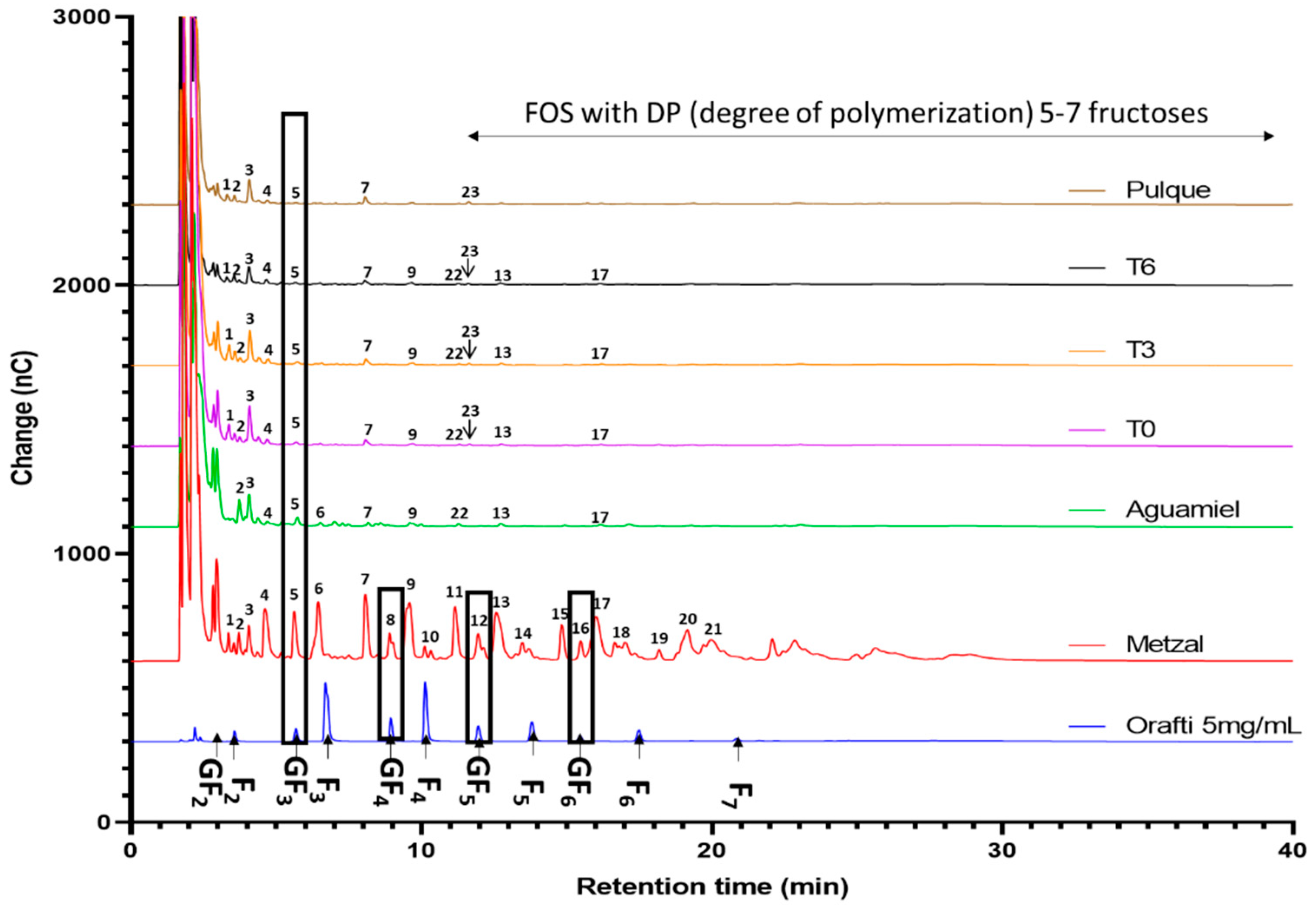
2.5. Fructooligosaccharides Identification
2.6. 16S rRNA Gene V3–V4 Fragment Amplicon and ITSR1 Library Preparation and Sequencing
2.7. Bioinformatics and Statistical Analysis
3. Results
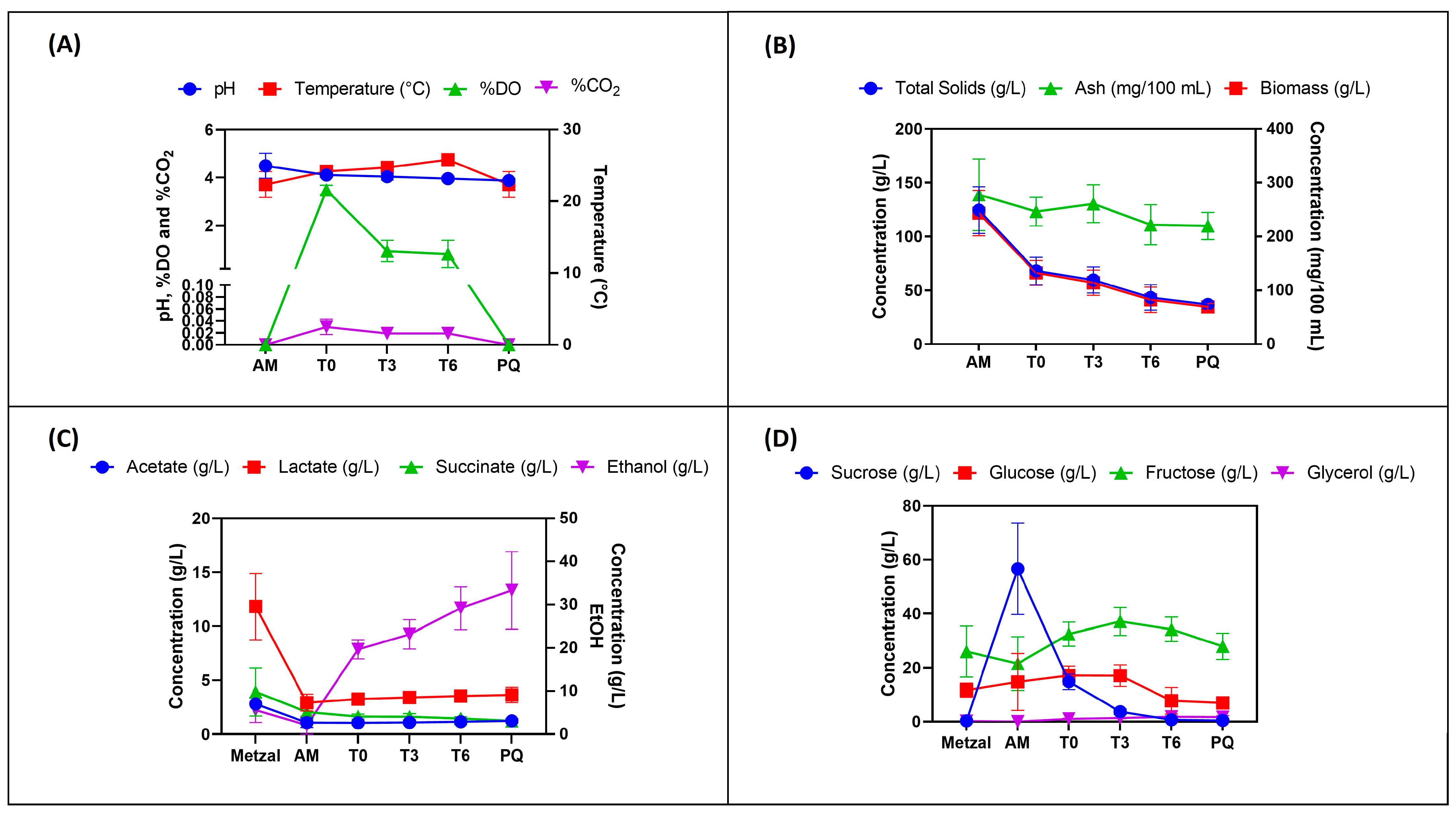
3.1. Physical Parameters of Sampled AM, PQ, and Laboratory Fermentation
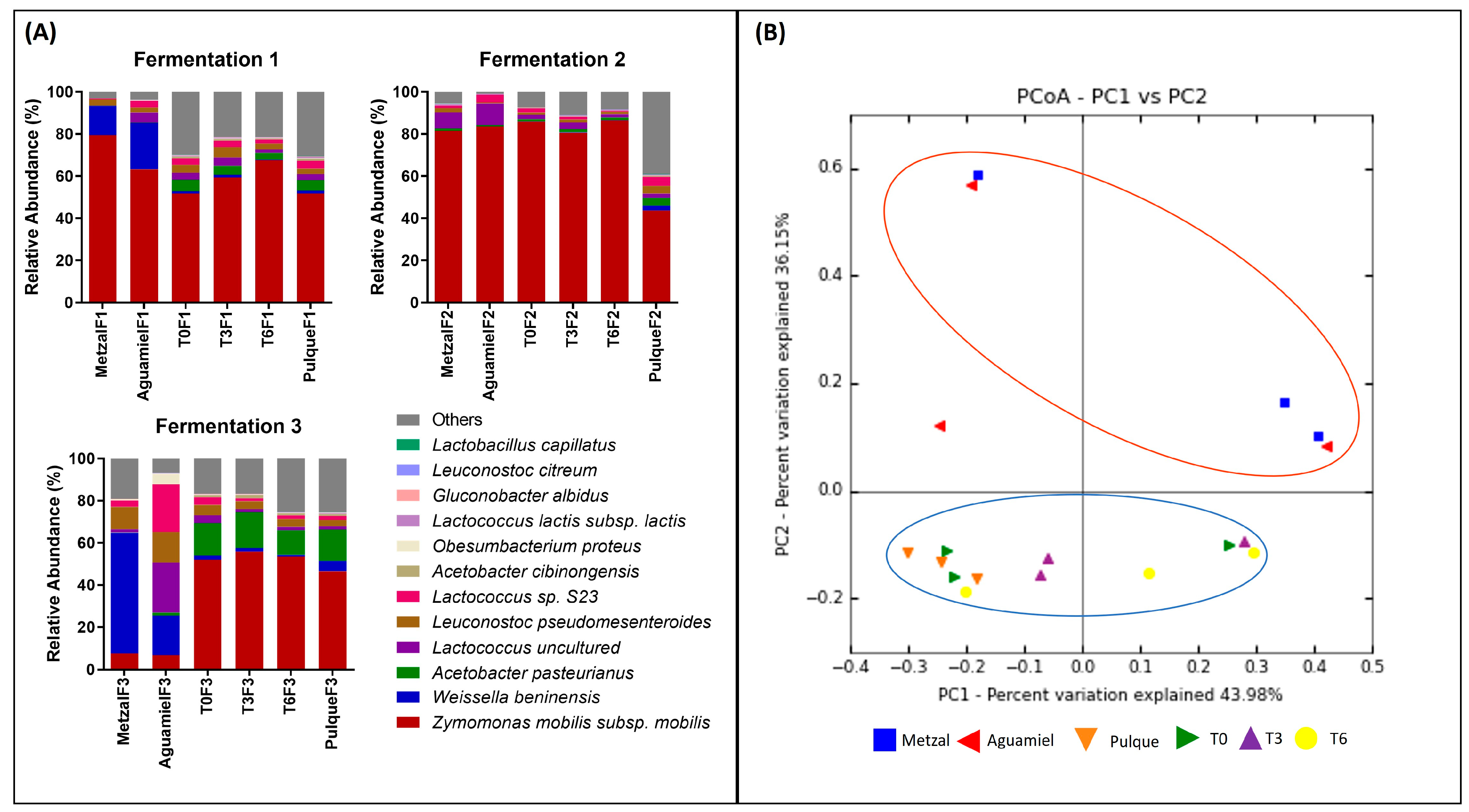
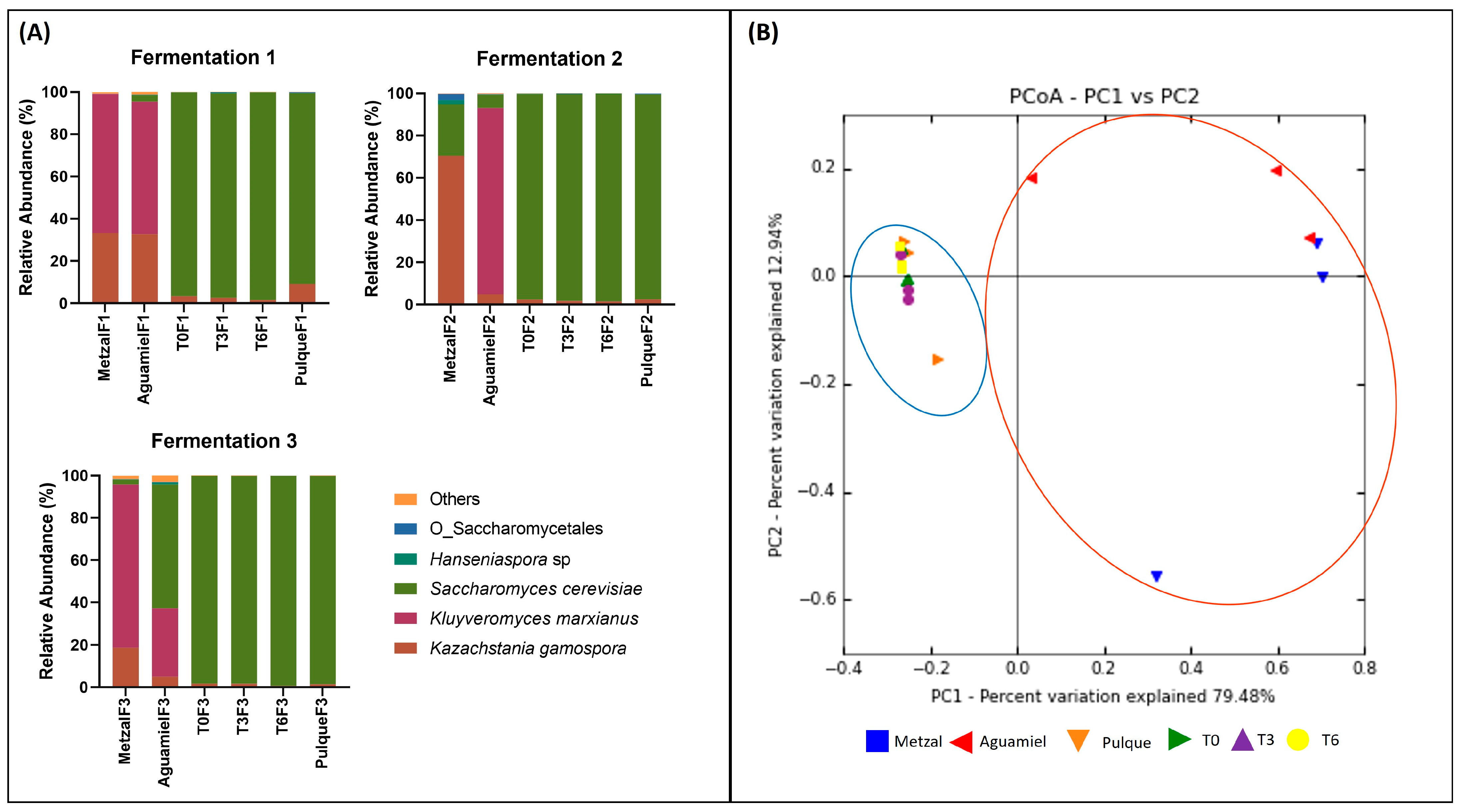
3.2. Total DNA Extraction and Sequencing and Taxonomy Assignment
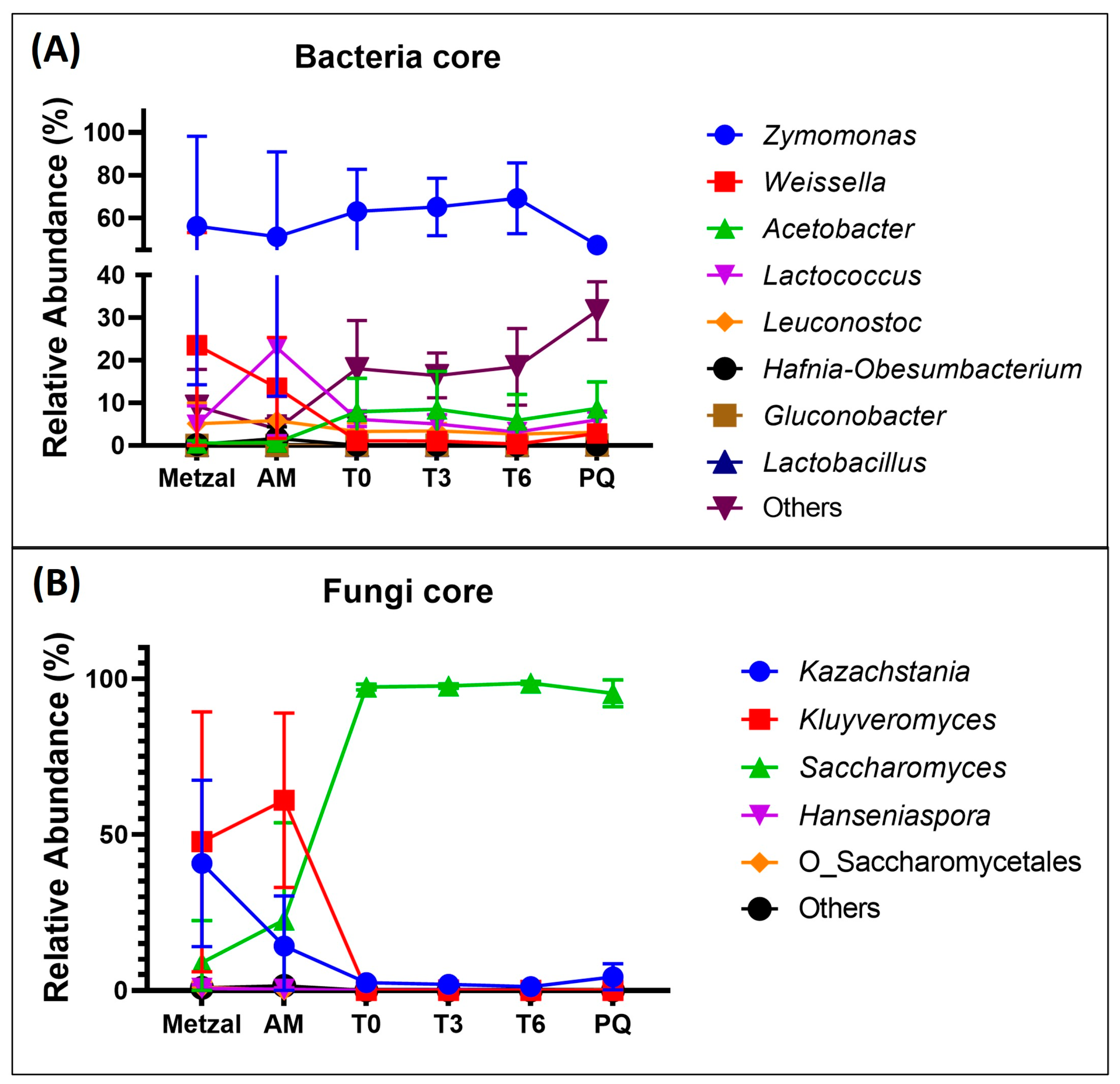
3.3. Taxonomy Assignment and Diversity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Escalante, A.; López Soto, D.R.; Velázquez Gutiérrez, J.E.; Giles-Gómez, M.; Bolívar, F.; López-Munguía, A. Pulque, a traditional Mexican alcoholic fermented beverage: Historical, microbiological, and technical aspects. Front. Microbiol. 2016, 7, 1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivieso Solís, D.G.; Vargas Escamilla, C.A.; Mondragón Contreras, N.; Galván Valle, G.A.; Gilés-Gómez, M.; Bolívar, F.; Escalante, A. Sustainable production of pulque and maguey in Mexico: Current situation and perspectives. Front. Sustain. Food Syst. 2021, 5, 678168. [Google Scholar] [CrossRef]
- Álvarez-Ríos, G.D.; Figueredo-Urbina, C.J.; Casas, A. Sistemas de manejo de maguey pulquero en México. Etnobiología 2020, 18, 3–23. [Google Scholar]
- Moreno-Terrazas, R.; Escalante, A.; Verdugo-Valdez, A.G.; De la Rosa, M.; Ortiz Basurto, R.I.; Orantes-García, C.; Palafox-González, L.; Castro Díaz, A.S.; Lappe-Oliveras, P. Bebidas refrescantes y alcohólicas de Agave. In Panorama Aprovechamiento de Agaves en México; CONACYT, CIATEJ, AGARED: Jalisco, México, 2017; pp. 123–163. ISBN 978-607-97548-2-2. [Google Scholar]
- Parsons, J.R.; Darling, J.A. Maguey (Agave spp.) utilization in Mesoamerican civilization: A case for Precolumbian “pastoralism”. Bol. Soc. Bot. Mex. 2000, 66, 81–91. [Google Scholar]
- Correa-Ascencio, M.; Robertson, I.G.; Cabrera-Cortes, O.; Cabrera-Castro, R.; Evershed, R.P. Pulque production from fermented Agave sap as a dietary supplement in prehispanic Mesoamerica. Proc. Natl. Acad. Sci. USA 2014, 111, 14223–14228. [Google Scholar] [CrossRef] [Green Version]
- Ramírez Rancaño, M. El rey del Pulque: Ignacio Torres Adalid y la Industria Pulquera; Plaza y Valdes; UNAM, Instituto de Investigaciones Sociales: Ciudad de México, México, 2000; ISBN 968-856-812-0. [Google Scholar]
- Gonçalves de Lima, O. El Maguey y el Pulque en los Codices Mexicanos; Fondo de Cultura Económica: Ciudad de México, México, 1956. [Google Scholar]
- Sánchez-Marroquín, A.; Terán, J.; Piso, J. Estudios sobre la microbiología del pulque—XVIII—Datos químicos de la fermentación de aguamiel con cultivos puros. Rev. Soc. Quím. Mex. 1957, 1, 167–174. [Google Scholar]
- Escalante, A.; Giles-Gómez, M.; Hernandez, G.; Cordova-Aguilar, M.; Lopez-Munguia, A.; Gosset, G.; Bolivar, F. Analysis of bacterial community during the fermentation of pulque, a traditional Mexican alcoholic beverage, using a polyphasic approach. Int. J. Food Microbiol. 2008, 124, 126–134. [Google Scholar] [CrossRef]
- Chacón-Vargas, K.; Torres, J.; Giles-Gómez, M.; Escalante, A.; Gibbons, J.G. Genomic profiling of bacterial and fungal communities and their predictive functionality during pulque fermentation by whole-genome shotgun sequencing. Sci. Rep. 2020, 10, 15115. [Google Scholar] [CrossRef]
- Rocha-Arriaga, C.; Espinal-Centeno, A.; Martinez-Sánchez, S.; Caballero-Pérez, J.; Alcaraz, L.D.; Cruz-Ramírez, A. Deep microbial community profiling along the fermentation process of pulque, a biocultural resource of Mexico. Microbiol. Res. 2020, 241, 126593. [Google Scholar] [CrossRef]
- Peralta-García, I.; González-Muñoz, F.; Elena, R.-A.M.; Sánchez-Flores, A.; López Munguía, A. Evolution of fructans in aguamiel (Agave sap) during the plant production lifetime. Front. Nutr. 2020, 7, 566950. [Google Scholar] [CrossRef]
- Ortiz-Basurto, R.I.; Pourcelly, G.; Doco, T.; Williams, P.; Dornier, M.; Belleville, M.-P. Analysis of the main components of the aguamiel produced by the maguey-pulquero (Agave mapisaga) throughout the harvest period. J. Agric. Food Chem. 2008, 56, 3682–3687. [Google Scholar] [CrossRef] [PubMed]
- del C. Martínez-Rodríguez, J.; De la Mora-Amutio, M.; Plascencia-Correa, L.A.; Audelo-Regalado, E.; Guardado, F.R.; Hernández-Sánchez, E.; Peña-Ramírez, Y.J.; Escalante, A.; Beltrán-García, M.J.; Ogura, T. Cultivable endophytic bacteria from leaf bases of Agave tequilana and their role as plant growth promoters. Braz. J. Microbiol. 2014, 45, 1333–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escalante, A.; Wacher, C.; Farrés, A. Lactic acid bacterial diversity in the traditional Mexican fermented dough pozol as determined by 16s rDNA sequence analysis. Int. J. Food Microbiol. 2001, 64, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Astudillo-Melgar, F.; Ochoa-Leyva, A.; Utrilla, J.; Huerta-Beristain, G. Bacterial diversity and population dynamics during the fermentation of palm wine from Guerrero Mexico. Front. Microbiol. 2019, 10, 531. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Kõljalg, U.; Nilsson, H.R.; Schigel, D.; Tedersoo, L.; Larsson, K.-H.; May, T.W.; Taylor, A.F.S.; Jeppesen, T.S.; Frøslev, T.G.; Lindahl, B.D.; et al. The Taxon hypothesis paradigm on the unambiguous detection and communication of taxa. Microorganisms 2020, 8, 1910. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-High-Throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Navas-Molina, J.A.; Peralta-Sánchez, J.M.; González, A.; McMurdie, P.J.; Vázquez-Baeza, Y.; Xu, Z.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J.; et al. Advancing our understanding of the human microbiome using QIIME. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 531, pp. 371–444. ISBN 978-0-12-407863-5. [Google Scholar]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. CP Bioinform. 2011, 36, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Marroquín, A.; Hope, P.H. Agave juice, fermentation and chemical composition studies of some species. J. Agric. Food Chem. 1953, 1, 246–249. [Google Scholar] [CrossRef]
- Escobar-Zepeda, A.; Montor, J.J.; Olvera, C.; Sanchez-Flores, A.; Lopez-Munguia, A. An extended taxonomic profile and metabolic potential analysis of pulque microbial community using metagenomics. J. Food Sci. Technol. 2020, 5, 83–97. [Google Scholar]
- Escalante, A.; Rodriguez, M.E.; Martinez, A.; López-Munguía, A.; Bolivar, F.; Gosset, G. Characterization of bacterial diversity in pulque, a traditional Mexican alcoholic fermented beverage, as determined by 16S rDNA analysis. FEMS Microbiol. Lett. 2004, 235, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadez-Blanco, R.; Bravo-Villa, G.; Santos-Sánchez, N.F.; Velasco-Almendarez, S.I.; Montville, T.J. The artisanal production of pulque, a traditional beverage of the Mexican highlands. Probiotics Antimicrob. Proteins 2012, 4, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Villarreal Morales, S.L.; Enríquez Salazar, M.I.; Michel Michel, M.R.; Flores Gallegos, A.C.; Montañez-Saens, J.; Aguilar, C.N.; Herrera, R.R. Metagenomic microbial diversity in aguamiel from two Agave species during 4-year seasons. Food Biotechnol. 2019, 33, 1–16. [Google Scholar] [CrossRef]
- Swings, J.; De Ley, J. The biology of Zymomonas. Bacteriol. Rev. 1977, 41, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Sahm, H.; Bringer-Maeyer, S.; Sprenger, G.A. The genus Zymomomonas. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 201–221. ISBN 978-0-387-25495-1. [Google Scholar]
- Weir, P.M. The ecology of Zymomonas: A review. Folia Microbiol. 2016, 61, 385–392. [Google Scholar] [CrossRef]
- Yang, S.; Fei, Q.; Zhang, Y.; Contreras, L.M.; Utturkar, S.M.; Brown, S.D.; Himmel, M.E.; Zhang, M. Zymomonas mobilis as a model system for production of biofuels and biochemicals. Microbiol. Biotechnol. 2016, 9, 699–717. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.; Stewart, G. Saccharomyces cerevisiae in the production of fermented beverages. Beverages 2016, 2, 30. [Google Scholar] [CrossRef]
- Jansen, M.L.A.; Bracher, J.M.; Papapetridis, I.; Verhoeven, M.D.; de Bruijn, H.; de Waal, P.P.; van Maris, A.J.A.; Klaassen, P.; Pronk, J.T. Saccharomyces cerevisiae strains for second-generation ethanol production: From academic exploration to industrial implementation. FEMS Yeast Res. 2017, 17, 1–20. [Google Scholar] [CrossRef]
- Chellapandian, M.; Larios, C.; Sanchez-Gonzalez, M.; Lopez-Munguia, A. Production and properties of a dextransucrase from Leuconostoc mesenteroides IBT-PQ isolated from ‘pulque’, a traditional Aztec alcoholic beverage. J. Ind. Microbiol. Biotechnol. 1998, 21, 51–56. [Google Scholar] [CrossRef]
- Dols, M.; Chraibi, W.; Remaud-Simeon, M.; Lindley, N.D.; Monsan, P.F. Growth and energetics of Leuconostoc mesenteroides NRRL B-1299 during metabolism of various sugars and their consequences for dextransucrase production. Appl. Environ. Microbiol. 1997, 63, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.M.; Zannini, E.; Wilkinson, S.; Daenen, L.; Arendt, E.K. Physiology of acetic acid bacteria and their role in vinegar and fermented beverages. Compr. Rev. Food Sci. Food Saf. 2019, 18, 587–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pothakos, V.; Illeghems, K.; Laureys, D.; Spitaels, F.; Vandamme, P.; De Vuyst, L. Acetic acid bacteria in fermented food and beverage ecosystems. In Acetic Acid Bacteria; Matsushita, K., Toyama, H., Tonouchi, N., Okamoto-Kainuma, A., Eds.; Springer: Tokyo, Japan, 2016; pp. 73–99. ISBN 978-4-431-55931-3. [Google Scholar]
- Ortiz-Soto, M.E.; Olivares-Illana, V.; López-Munguía, A. Biochemical properties of inulosucrase from Leuconostoc citreum CW28 used for inulin synthesis. Biocatal. Biotransform. 2004, 22, 275–281. [Google Scholar] [CrossRef]
- Lappe-Oliveras, P.; Moreno-Terrazas, R.; Arrizón-Gaviño, J.; Herrera-Suárez, T.; García-Mendoza, A.; Gschaedler-Mathis, A. Yeasts associated with the production of Mexican alcoholic nondistilled and distilled Agave beverages. FEMS Yeast Res. 2008, 8, 1037–1052. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Guerrero, A.E.; Olvera, J.L.; García-Garibay, M.; Gómez-Ruiz, L. Inulinase-hyperproducing strains of Kluyveromyces sp. isolated from aguamiel (Agave Sap) and pulque. World J. Microbiol. Biotechnol. 2005, 22, 115–117. [Google Scholar] [CrossRef]
- Karim, A.; Gerliani, N.; Aïder, M. Kluyveromyces marxianus: An emerging yeast cell factory for applications in food and biotechnology. Int. J. Food Microbiol. 2020, 333, 108818. [Google Scholar] [CrossRef]
- Ha-Tran, D.M.; Nguyen, T.T.M.; Huang, C.-C. Kluyveromyces marxianus: Current state of omics studies, strain improvement strategy and potential industrial implementation. Fermentation 2020, 6, 124. [Google Scholar] [CrossRef]
- Michel-Cuello, C.; Gallegos-Fonseca, G.; Maldonado-Cervantes, E.; Aguilar-Rivera, N. Efecto de la temperatura y el pH del medio en la hidrólisis de fructanos de maguey para la obtención de jarabes fructosados. Rev. Mex. Ing. Quím. 2015, 14. [Google Scholar]
- Vallejo-García, L.C.; Rodríguez-Alegría, M.E.; López Munguía, A. Enzymatic process yielding a diversity of inulin-type microbial fructooligosaccharides. J. Agric. Food Chem. 2019, 67, 10392–10400. [Google Scholar] [CrossRef]
- Rodríguez-Alegría, M.E.; Enciso-Rodríguez, A.; Ortiz-Soto, M.E.; Cassani, J.; Olvera, C.; Munguía, A.L. Fructooligosaccharide production by a truncated Leuconostoc citreum inulosucrase mutant. Biocatal. Biotransform. 2010, 28, 51–59. [Google Scholar] [CrossRef]
- Torres-Rodríguez, I.; Rodríguez-Alegría, M.E.; Miranda-Molina, A.; Giles-Gómez, M.; Morales, R.C.; López-Munguía, A.; Bolívar, F.; Escalante, A. Screening and characterization of extracellular polysaccharides produced by Leuconostoc kimchii isolated from traditional fermented pulque beverage. SpringerPlus 2014, 3, 583. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Sample 1 | Sample 2 | Sample 3 |
|---|---|---|---|
| Date | 7 November 2019 | 11 November 2019 | 14 November 2019 |
| Environmental temperature | 22 °C | 21 °C | 20 °C |
| Environmental humidity | 87% | 89% | 88% |
| Ambient pressure | 732 atm | 731 atm | 731 atm |
| Weather | Foggy | Sunny | Cloudy |
| Diversity Index/Sample | Observed_OTUs | Chao1 | Shannon–Wiener | Simpson |
|---|---|---|---|---|
| Bacteria | ||||
| Metzal n = 3 | 77.0 ± 20.66 | 126.22 ± 31.11 | 1.56 ± 0.61 | 0.44 ± 0.17 |
| Aguamiel n = 3 | 60.67 ± 17.93 | 106.37 ± 38.66 | 1.96 ± 1.06 | 0.56 ± 0.28 |
| T0 n = 3 | 80.33 ± 9.71 | 128.17 ± 23.22 | 2.25 ± 0.96 | 0.56 ± 0.25 |
| T3 n = 3 | 99.67 ± 18.48 | 146.29 ± 43.47 | 2.24 ± 0.62 | 0.55 ± 0.17 |
| T6 n = 3 | 86.33 ± 15.50 | 161.53 ± 63.55 | 1.97 ± 0.77 | 0.49 ± 0.22 |
| Pulque n = 3 | 96.67 ± 22.81 | 156.46 ± 60.47 | 3.06 ± 0.18 | 0.75 ± 0.03 |
| Fungi | ||||
| Metzal n = 3 | 15.66 ± 3.78 | 16.77 ± 5.39 | 1.09 ± 0.15 | 0.442 ± 0.04 * |
| Aguamiel n = 3 | 20.66 ± 3.51 | 21.66 ± 2.08 ** | 1.26 ± 0.55 | 0.44 ± 0.20 |
| T0 n = 3 | 12.33 ± 0.57 | 12.33 ± 0.57 | 0.68 ± 0.03 | 0.20 ± 0.008 |
| T3 n = 3 | 13.00 ± 0.00 | 13.00 ± 0.00 | 0.65 ± 0.03 | 0.19 ± 0.01 |
| T6 n = 3 | 13.00 ± 1.00 | 13.50 ± 1.80 | 0.61 ± 0.02 * | 0.18 ± 0.002 * |
| Pulque n = 3 | 13.33 ± 1.15 | 13.33 ± 1.15 | 0.77 ± 0.16 | 0.24 ± 0.06 |
| Core | Relative Abundance (%) | |
|---|---|---|
| Bacteria (Genus level) | Zymomonas | 58.76 |
| Lactococcus | 8.04 | |
| Weissella | 7.08 | |
| Acetobacter | 5.4 | |
| Leuconostoc | 3.91 | |
| Obesumbacterium | 0.38 | |
| Gluconobacter | 0.06 | |
| Lactobacillus | 0.05 | |
| Total | 83.68 | |
| Fungi | Saccharomyces cerevisiae | 70.07 |
| Kluyveromyces marxianus | 18.13 | |
| Kazachstania gamospora | 10.84 | |
| Hanseniaspora sp. | 0.27 | |
| O_Saccharomycetales | 0.27 | |
| Total | 99.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Astudillo-Melgar, F.; Hernández-Chávez, G.; Rodríguez-Alegría, M.E.; Bolívar, F.; Escalante, A. Analysis of the Microbial Diversity and Population Dynamics during the Pulque Fermentation Process. Fermentation 2023, 9, 342. https://doi.org/10.3390/fermentation9040342
Astudillo-Melgar F, Hernández-Chávez G, Rodríguez-Alegría ME, Bolívar F, Escalante A. Analysis of the Microbial Diversity and Population Dynamics during the Pulque Fermentation Process. Fermentation. 2023; 9(4):342. https://doi.org/10.3390/fermentation9040342
Chicago/Turabian StyleAstudillo-Melgar, Fernando, Georgina Hernández-Chávez, María Elena Rodríguez-Alegría, Francisco Bolívar, and Adelfo Escalante. 2023. "Analysis of the Microbial Diversity and Population Dynamics during the Pulque Fermentation Process" Fermentation 9, no. 4: 342. https://doi.org/10.3390/fermentation9040342