
A Toolkit for Effective and Successive Genome Engineering of Escherichia coli

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, Editing Template, and Oligonucleotides
2.2. Competent Cells Preparation and Transformations
2.3. Media and Bacterial Cell Cultivation
2.4. PHBV Extraction and Analysis
2.5. Statistical Analysis
3. Results
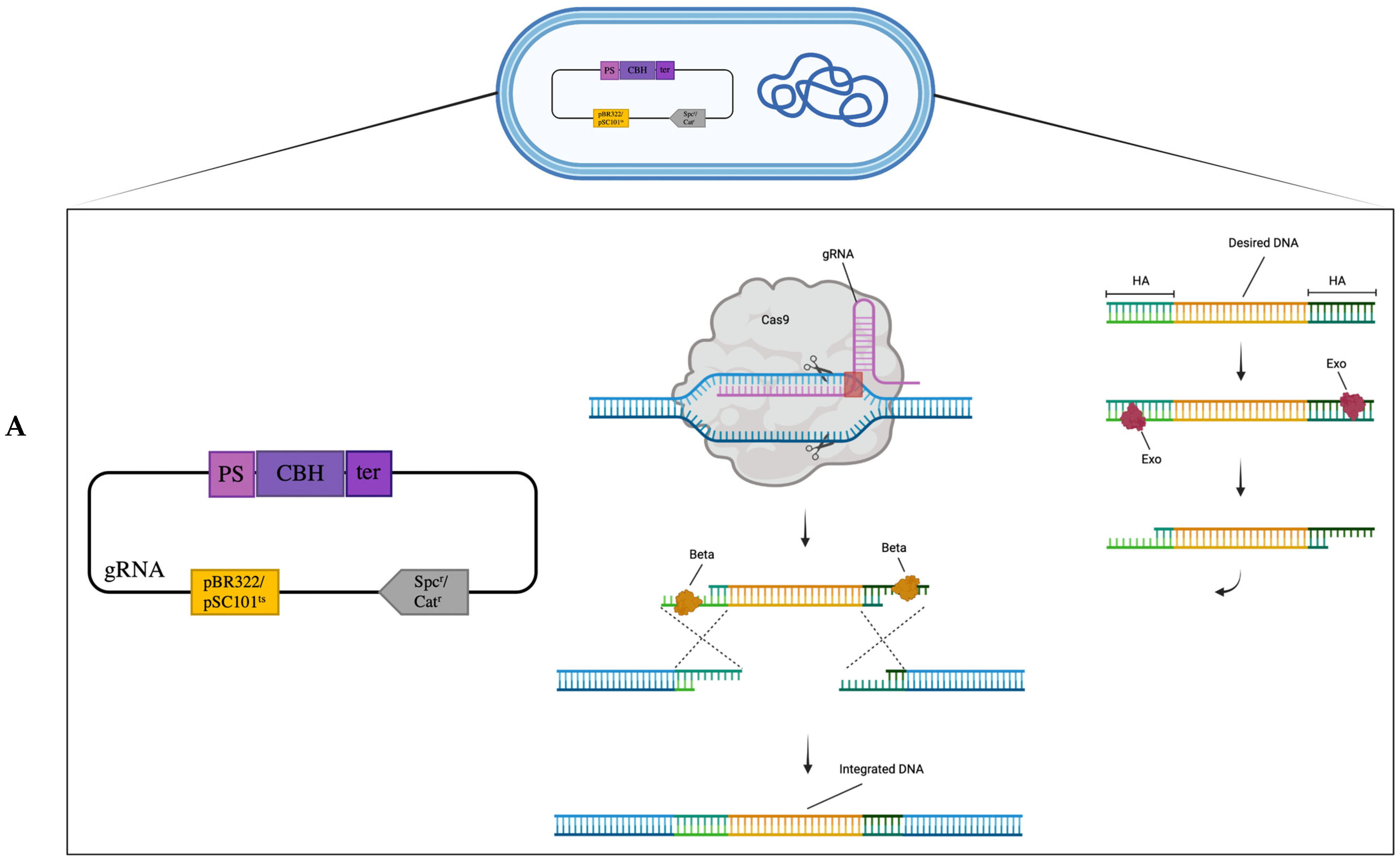
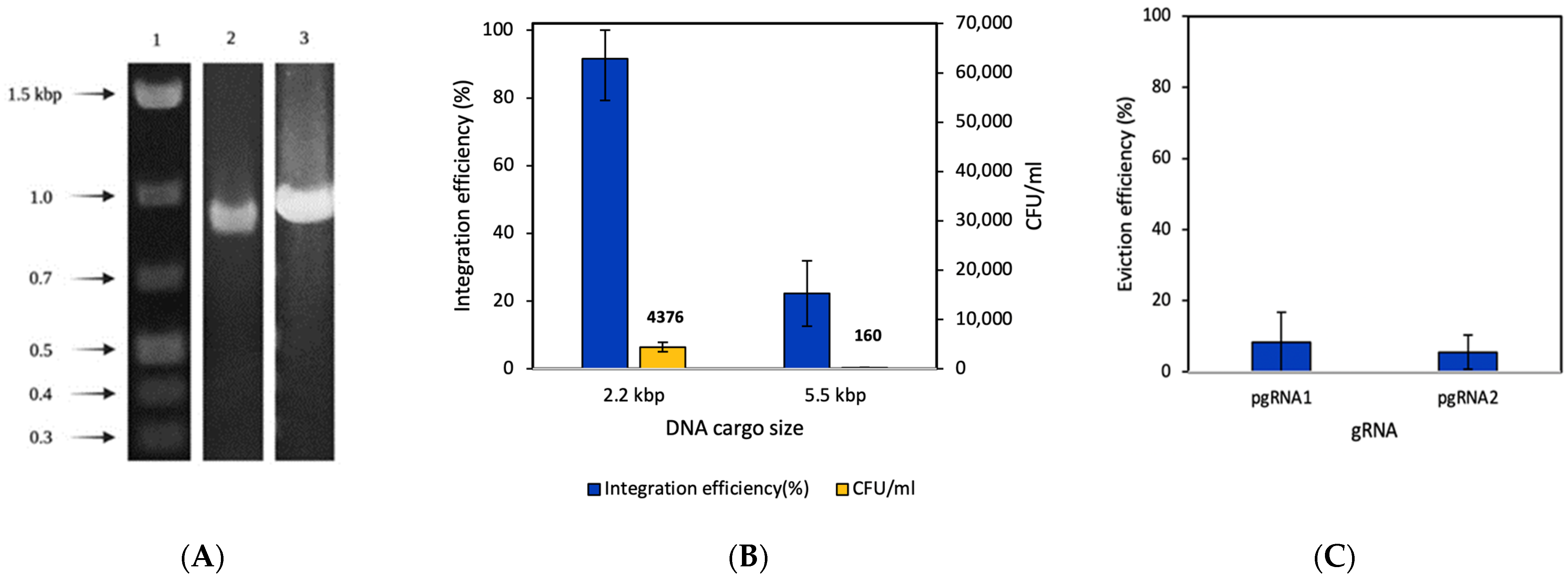
3.1. Genomic Knock-In Based on CRISPR-Cas9 Coupled with λRed Recombineering
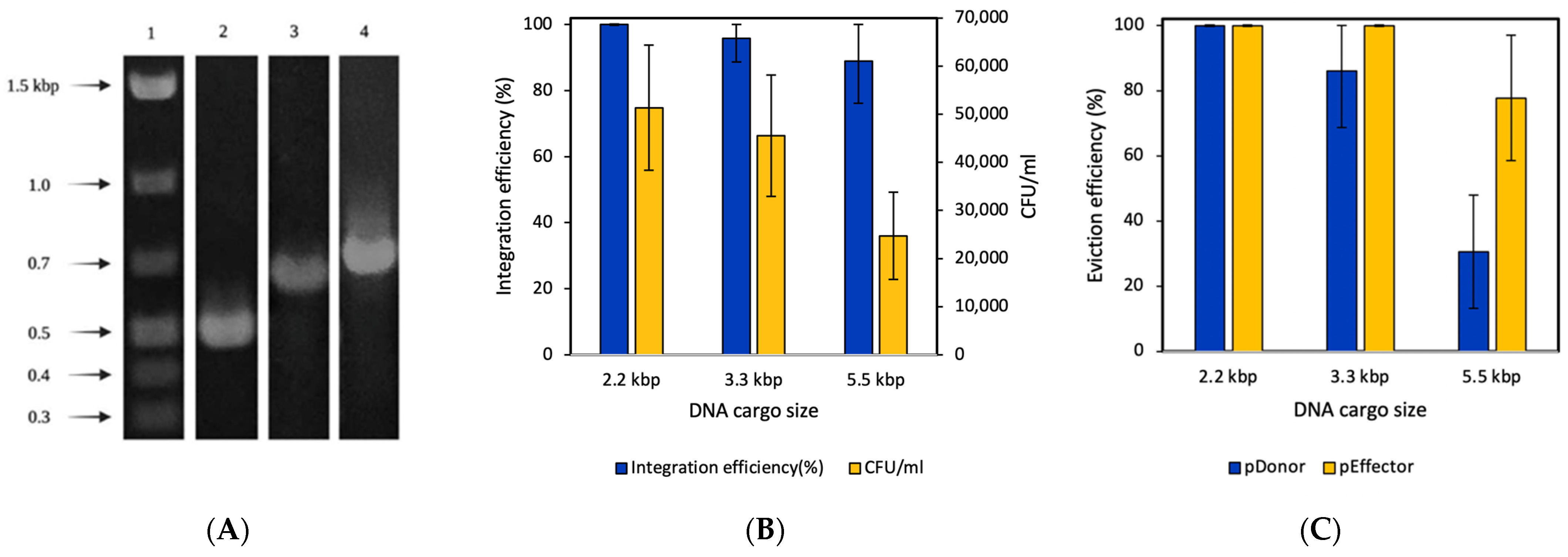
3.2. Genomic Knock-In Based on Transposon-Associated CRISPR-Cas System
3.3. Gene Knockout Based on CRISPR-Cas9 Coupled with λRed Recombineering
3.4. PHBV Biosynthesis Using Genome-Engineered Strains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Theisen, M.; Liao, J. Industrial biotechnology: Escherichia coli as a host. Ind. Biotechnol. Microorg. 2017, 1, 149–181. [Google Scholar]
- Zhang, Y.; Sun, X.; Wang, Q.; Xu, J.; Dong, F.; Yang, S.; Yang, J.; Zhang, Z.; Qian, Y.; Chen, J. Multicopy chromosomal integration using CRISPR-associated transposases. ACS Synth. Biol. 2020, 9, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Fenster, J.A.; Fankhauser, R.G.; Kaar, J.L.; Tenaillon, O.; Gill, R.T. CRISPR/Cas9 recombineering-mediated deep mutational scanning of essential genes in Escherichia coli. Mol. Syst. Biol. 2020, 16, e9265. [Google Scholar] [CrossRef] [PubMed]
- Bassalo, M.C.; Garst, A.D.; Halweg-Edwards, A.L.; Grau, W.C.; Domaille, D.W.; Mutalik, V.K.; Arkin, A.P.; Gill, R.T. Rapid and efficient one-step metabolic pathway integration in E. coli. ACS Synth. Biol. 2016, 5, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, X.-Y.; Du, H.-T.; Zhang, X.; Ma, Y.-M.; Chen, J.-C.; Ye, J.-W.; Jiang, X.-R.; Chen, G.-Q. Chromosome engineering of the TCA cycle in Halomonas bluephagenesis for production of copolymers of 3-hydroxybutyrate and 3-hydroxyvalerate (PHBV). Metab. Eng. 2019, 54, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, V.C.; Zeiger, E.; Brusick, D.; McCoy, E.; McGregor, D.; Mortelmans, K.; Rosenkranz, H.S.; Simmon, V.F. Reproducibility of microbial mutagenicity assays: II. Testing of carcinogens and noncarcinogens in Salmonella typhimurium and Escherichia coli. Environ. Mutagen. 1985, 7, 1–19. [Google Scholar] [CrossRef]
- Witkin, E.M. Ultraviolet mutagenesis and inducible DNA repair in Escherichia coli. Bacteriol. Rev. 1976, 40, 869–907. [Google Scholar] [CrossRef]
- Smith, J.; Bibikova, M.; Whitby, F.G.; Reddy, A.; Chandrasegaran, S.; Carroll, D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000, 28, 3361–3369. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Horii, Z.-I.; Clark, A. Genetic analysis of the recF pathway to genetic recombination in Escherichia coli K12: Isolation and characterization of mutants. J. Mol. Biol. 1973, 80, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Isaacs, F.J.; Carr, P.A.; Sun, Z.Z.; Xu, G.; Forest, C.R.; Church, G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, R.; Priefer, U.; Pühler, A. A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in gram negative bacteria. Bio/technology 1983, 1, 784–791. [Google Scholar] [CrossRef]
- Morse, M.; Lederberg, E.; Lederberg, J. Transduction in Escherichia coli K-12. Genetics 1956, 41, 142. [Google Scholar] [CrossRef] [PubMed]
- Llosa, M.; de la Cruz, F. Bacterial conjugation: A potential tool for genomic engineering. Res. Microbiol. 2005, 156, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wannier, T.M.; Nyerges, A.; Kuchwara, H.M.; Czikkely, M.; Balogh, D.; Filsinger, G.T.; Borders, N.C.; Gregg, C.J.; Lajoie, M.J.; Rios, X. Improved bacterial recombineering by parallelized protein discovery. Proc. Natl. Acad. Sci. USA 2020, 117, 13689–13698. [Google Scholar] [CrossRef] [PubMed]
- Mosberg, J.; Lajoie, M.; Church, G. Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics 2010, 186, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.K.; Stahl, F.W. Interaction between the sbcC gene of Escherichia coli and the gam gene of phage lambda. Genetics 1989, 123, 249–253. [Google Scholar] [CrossRef]
- Sawitzke, J.A.; Thomason, L.C.; Costantino, N.; Bubunenko, M.; Datta, S.; Court, D.L. Recombineering: In vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol. 2007, 421, 171–199. [Google Scholar]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, P.L.H.; Ronda, C.; Klompe, S.E.; Chen, E.E.; Acree, C.; Wang, H.H.; Sternberg, S.H. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat. Biotechnol. 2021, 39, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, H.; Lee, S. CRISPR-Cas9-mediated pinpoint microbial genome editing aided by target-mismatched sgRNAs. Genome Res. 2020, 30, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Petassi, M.T.; Hsieh, S.-C.; Peters, J. Guide RNA categorization enables target site choice in Tn7-CRISPR-Cas transposons. Cell 2020, 183, 1757–1771.e18. [Google Scholar] [CrossRef]
- Fokum, E.; Zabed, H.M.; Guo, Q.; Yun, J.; Yang, M.; Pang, H.; An, Y.; Li, W.; Qi, X. Metabolic engineering of bacterial strains using CRISPR/Cas9 systems for biosynthesis of value-added products. Food Biosci. 2019, 28, 125–132. [Google Scholar] [CrossRef]
- Li, Y.; Glass, Z.; Huang, M.; Chen, Z.-Y.; Xu, Q. Ex vivo cell-based CRISPR/Cas9 genome editing for therapeutic applications. Biomaterials 2020, 234, 119711. [Google Scholar] [CrossRef]
- Lin, C.S.; Hsu, C.T.; Yang, L.H.; Lee, L.Y.; Fu, J.Y.; Cheng, Q.W.; Wu, F.H.; Hsiao, H.C.W.; Zhang, Y.; Zhang, R. Application of protoplast technology to CRISPR/Cas9 mutagenesis: From single-cell mutation detection to mutant plant regeneration. Plant Biotechnol. J. 2018, 16, 1295–1310. [Google Scholar] [CrossRef] [Green Version]
- Klompe, S.E.; Vo, P.L.; Halpin-Healy, T.S.; Sternberg, S.H. Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature 2019, 571, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Ladha, A.; Strecker, J.; Faure, G.; Neumann, E.; Altae-Tran, H.; Macrae, R.K.; Zhang, F. Dual modes of CRISPR-associated transposon homing. Cell 2021, 184, 2441–2453.e18. [Google Scholar] [CrossRef] [PubMed]
- Strecker, J.; Ladha, A.; Gardner, Z.; Schmid-Burgk, J.L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019, 365, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Halpin-Healy, T.S.; Klompe, S.E.; Sternberg, S.H.; Fernández, I.S. Structural basis of DNA targeting by a transposon-encoded CRISPR–Cas system. Nature 2020, 577, 271–274. [Google Scholar] [CrossRef]
- van der Oost, J. IMougiakos, First structural insights into CRISPR-Cas-guided DNA transposition. Cell Res. 2020, 30, 193–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Xu, W.; Yang, H. Structural basis of a Tn7-like transposase recruitment and DNA loading to CRISPR-Cas surveillance complex. Cell Res. 2020, 30, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Vo, P.L.H.; Acree, C.; Smith, M.L.; Sternberg, S.H. Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. Mob. DNA 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Rodríguez, L.T.; Ellington, A.; Reisch, C. Broad-host-range mutagenesis with CRISPR-associated transposase. bioRxiv 2022. [Google Scholar] [CrossRef]
- Yang, J.; Yang, J.; Zhang, Y.; Yang, S.; Zhang, J.; Jiang, Y.; Yang, S. CRISPR-Associated Transposase System Can Insert Multiple Copies of Donor DNA into the Same Target Locus. CRISPR J. 2021, 4, 789–798. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Y.; Xu, J.; Zhang, J.; Zhang, J.; Yang, J.; Jiang, Y.; Yang, S. Orthogonal CRISPR-associated transposases for parallel and multiplexed chromosomal integration. Nucleic Acids Res. 2021, 49, 10192–10202. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Amann, E.; Ochs, B.; Abel, K.-J. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 1988, 69, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W.; Sambrook, J. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001; Volume 1. [Google Scholar]
- Srirangan, K.; Liu, X.; Westbrook, A.; Akawi, L.; Pyne, M.E.; Moo-Young, M.; Chou, C.P. Biochemical, genetic, and metabolic engineering strategies to enhance coproduction of 1-propanol and ethanol in engineered Escherichia coli. Appl. Microbiol. Biotechnol. 2014, 98, 9499–9515. [Google Scholar] [CrossRef]
- Westbrook, A.W.; Moo-Young, M.; Chou, C. Development of a CRISPR-Cas9 tool kit for comprehensive engineering of Bacillus subtilis. Appl. Environ. Microbiol. 2016, 82, 4876–4895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherepanov, P.P.; Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1995, 158, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Miscevic, D.; Mao, J.-Y.; Mozell, B.; Srirangan, K.; Abedi, D.; Moo-Young, M.; Chou, C.P. Bio-based production of poly (3-hydroxybutyrate-co-3-hydroxyvalerate) with modulated monomeric fraction in Escherichia coli. Appl. Microbiol. Biotechnol. 2021, 105, 1435–1446. [Google Scholar] [CrossRef]
- Hiroe, A.; Tsuge, K.; Nomura, C.T.; Itaya, M.; Tsuge, T. Rearrangement of gene order in the phaCAB operon leads to effective production of ultrahigh-molecular-weight poly [(R)-3-hydroxybutyrate] in genetically engineered Escherichia coli. Appl. Environ. Microbiol. 2012, 78, 3177–3184. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Michisaka, S.; Nozaki, S.; Funabashi, T.; Hirano, T.; Takei, R.; Nakayama, K. Practical method for targeted disruption of cilia-related genes by using CRISPR/Cas9-mediated, homology-independent knock-in system. Mol. Biol. Cell 2017, 28, 898–906. [Google Scholar] [CrossRef]
- Sukhija, K.; Pyne, M.; Ali, S.; Orr, V.; Abedi, D.; Moo-Young, M.; Chou, C.P. Developing an extended genomic engineering approach based on recombineering to knock-in heterologous genes to Escherichia coli genome. Mol. Biotechnol. 2012, 51, 109–118. [Google Scholar] [CrossRef]
- Rivero-Müller, A.; Lajić, S.; Huhtaniemi, I. Assisted large fragment insertion by Red/ET-recombination (ALFIRE)—An alternative and enhanced method for large fragment recombineering. Nucleic Acids Res. 2007, 35, e78. [Google Scholar] [CrossRef] [Green Version]
- Pyne, M.E.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Coupling the CRISPR/Cas9 system with lambda red recombineering enables simplified chromosomal gene replacement in Escherichia coli. Appl. Environ. Microbiol. 2015, 81, 5103–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresca, M.; Erler, A.; Fu, J.; Friedrich, A.; Zhang, Y.; Stewart, A. Single-stranded heteroduplex intermediates in λ Red homologous recombination. BMC Mol. Biol. 2010, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, Z.; Huang, C.; Zhang, Y.; Wang, Z.; Tang, Y.-j.; Chen, T.; Zhao, X. Metabolic engineering of Escherichia coli using CRISPR–Cas9 meditated genome editing. Metab. Eng. 2015, 31, 13–21. [Google Scholar] [CrossRef]
- Cui, L.; Bikard, D. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 2016, 44, 4243–4251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, K.; Sato, Y.; Hizume, T.; Honda, K. Genome editing by miniature CRISPR/Cas12f1 enzyme in Escherichia coli. J. Biosci. Bioeng. 2021, 132, 120–124. [Google Scholar] [CrossRef]
- Srirangan, K.; Liu, X.; Tran, T.T.; Charles, T.C.; Moo-Young, M.; Chou, C.P. Engineering of Escherichia coli for direct and modulated biosynthesis of poly (3-hydroxybutyrate-co-3-hydroxyvalerate) copolymer using unrelated carbon sources. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Horng, Y.-T.; Chien, C.-C.; Huang, C.-T.; Wei, Y.-H.; Chen, S.-Y.; Lan, J.-W.; Soo, P.-C. Biosynthesis of poly (3-hydroxybutyrate-co-3-hydroxyvalerate) with co-expressed propionate permease (prpP), beta-ketothiolase B (bktB), and propionate-CoA synthase (prpE) in Escherichia coli. Biochem. Eng. J. 2013, 78, 73–79. [Google Scholar] [CrossRef]
- Yang, J.E.; Choi, Y.J.; Lee, S.J.; Kang, K.-H.; Lee, H.; Oh, Y.H.; Lee, S.H.; Park, S.J.; Lee, S.Y. Metabolic engineering of Escherichia coli for biosynthesis of poly (3-hydroxybutyrate-co-3-hydroxyvalerate) from glucose. Appl. Microbiol. Biotechnol. 2014, 98, 95–104. [Google Scholar] [CrossRef]
- Miscevic, D.; Mao, J.Y.; Moo-Young, M.; Chou, C.H.P. High-level heterologous production of propionate in engineered Escherichia coli. Biotechnol. Bioeng. 2020, 117, 1304–1315. [Google Scholar] [CrossRef]
- Yazdani, S.S.; Gonzalez, R. Anaerobic fermentation of glycerol: A path to economic viability for the biofuels industry. Curr. Opin. Biotechnol. 2007, 18, 213–219. [Google Scholar] [CrossRef]
- Mattam, A.J.; Clomburg, J.M.; Gonzalez, R.; Yazdani, S.S. Fermentation of glycerol and production of valuable chemical and biofuel molecules. Biotechnol. Lett. 2013, 35, 831–842. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description or Relevant Genotype | Source |
|---|---|---|
| E. coli host strains | ||
| HI-Control 10G | mcrA ∆ (mrr-hsdRMS-mcrBC) endA1 recA1 80dlacZ∆M15 ∆lacX74 araD139 ∆ (ara leu)7697 galU galK rpsL (Strr) nupG λ− tonA Mini-F lacIq1 (Gentr) | Lucigen |
| MG1655 | K-12; F− λ− rph-1 | Lab stock |
| CPC-Sbm | BWΔldhA, Ptrc::sbm (i.e., with the FRT-Ptrc cassette replacing the 204-bp up-stream of the Sbm operon) | Lab stock |
| ST001 | CPC-Sbm endA::(PBAD::::) | Lab stock |
| ST002 | CPC-Sbm endA::(PBAD::::) yjcS::(PtetA::spc.-gRNA.P279T-cas9) | This study |
| ST003 | CPC-Sbm endA::(PBAD::::) yjcS::(PtetA::spc.-gRNA.P279T-cas9) bcsA::(Pgracmax(T7.RBS)::bktB:phaB) | This study |
| ST004 | CPC-Sbm endA::(PBAD::::) yjcS::(PtetA::spc.-gRNA.P279T-cas9) bcsA::(Pgracmax(T7.RBS)::bktB:phaB) intF::(Pgracmax(T7.RBS)::phaC:phaA | This study |
| ST005 | ST004 ∆iclR | This study |
| ST006 | ST004 ∆iclR ∆sdhA | This study |
| ST007 | CPC-Sbm yjcS::(PtetA::spc.-gRNA.P279T-cas9) | This study |
| Plasmids | ||
| pDonor | Donor plasmid for V. Cholerae transposon system | [22] |
| pEffector | Effector expression for V. Cholerae transposon system | [22] |
| pUC19 | Subcloning plasmid for generating Effector plasmids | Lab stock |
| pTrc99a | pBR322 ori, Ampr | [42] |
| pSC101 | pSC101ts ori, Catr | Lab stock |
| pDonor1 | pBR322 ori, PtetA::spc.-gRNA.P279T-cas9, Spcr | This study |
| pDonor2 | pBR322 ori, Pgracmax(T7.RBS)::bktB:phaB, Spcr | This study |
| pDonor3 | pBR322 ori, Pgracmax(T7.RBS)::phaC:phaA, Spcr | This study |
| pEffector1 | pSC101ts ori, PJ23119::yjcS-gRNA.P882T, Catr | This study |
| pEffector2 | pSC101ts ori, PJ23119::bcsA-gRNA.P1249NT, Catr | This study |
| pEffector3 | pSC101ts ori, PJ23119::intF-gRNA.P242T, Catr | This study |
| pbktB.phaB-bcsA | pBR322 ori, Pgracmax(T7.RBS)::bktB:phaB, Ampr | This study |
| pCas9-yjcS | pBR322 ori, PtetA::spc.-gRNA.P279T-cas9, Ampr | This study |
| pgRNA1 | pBR322 ori, PxylA.SphI :: yjcS-gRNA.P1032T, Spcr | This study |
| pgRNA2 | pBR322 ori, PxylA.SphI :: bcsA-gRNA.P1088T, Spcr | This study |
| pgRNA3 | pSC101ts ori, PxylA.SphI :: iclR-gRNA.P78T, Catr | This study |
| pgRNA4 | pSC101ts ori, PxylA.SphI :: sdhA-gRNA.P392T, Catr | This study |
| pCas9 | pSC101ts ori, PtetA::spc.-gRNA.P279T-cas9, Catr | Lab stock |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arab, B.; Westbrook, A.; Moo-Young, M.; Chou, C.-H.P. A Toolkit for Effective and Successive Genome Engineering of Escherichia coli. Fermentation 2023, 9, 14. https://doi.org/10.3390/fermentation9010014
Arab B, Westbrook A, Moo-Young M, Chou C-HP. A Toolkit for Effective and Successive Genome Engineering of Escherichia coli. Fermentation. 2023; 9(1):14. https://doi.org/10.3390/fermentation9010014
Chicago/Turabian StyleArab, Bahareh, Adam Westbrook, Murray Moo-Young, and Chih-Hsiung Perry Chou. 2023. "A Toolkit for Effective and Successive Genome Engineering of Escherichia coli" Fermentation 9, no. 1: 14. https://doi.org/10.3390/fermentation9010014