
Comparative Genome Analysis of Two Heterotrophic Nitrifying Pseudomonas putida Strains Isolated from Freshwater Shrimp Ponds in Soc Trang Province

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. DNA Extraction, Genome Sequencing and Annotation
2.2. QC and De Novo Assembly
2.3. Identity of Assembled Genomes
2.4. Annotation of Selected Genome Sequences
2.5. Phylogenetic Tree Analysis
2.6. Comparative Genome Analysis
2.7. Genomic Island Prediction
3. Results and Discussion
3.1. Genome Features
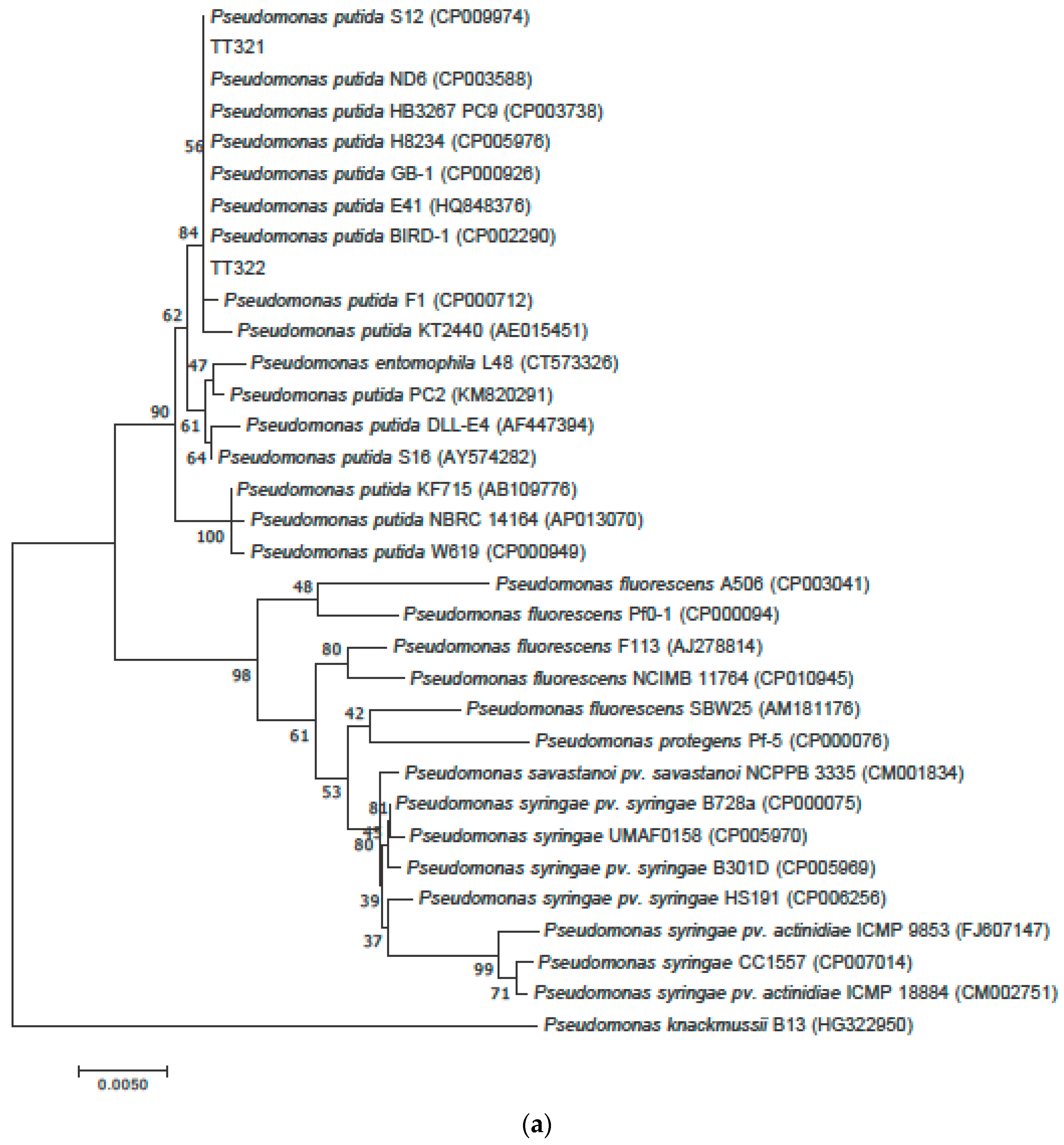
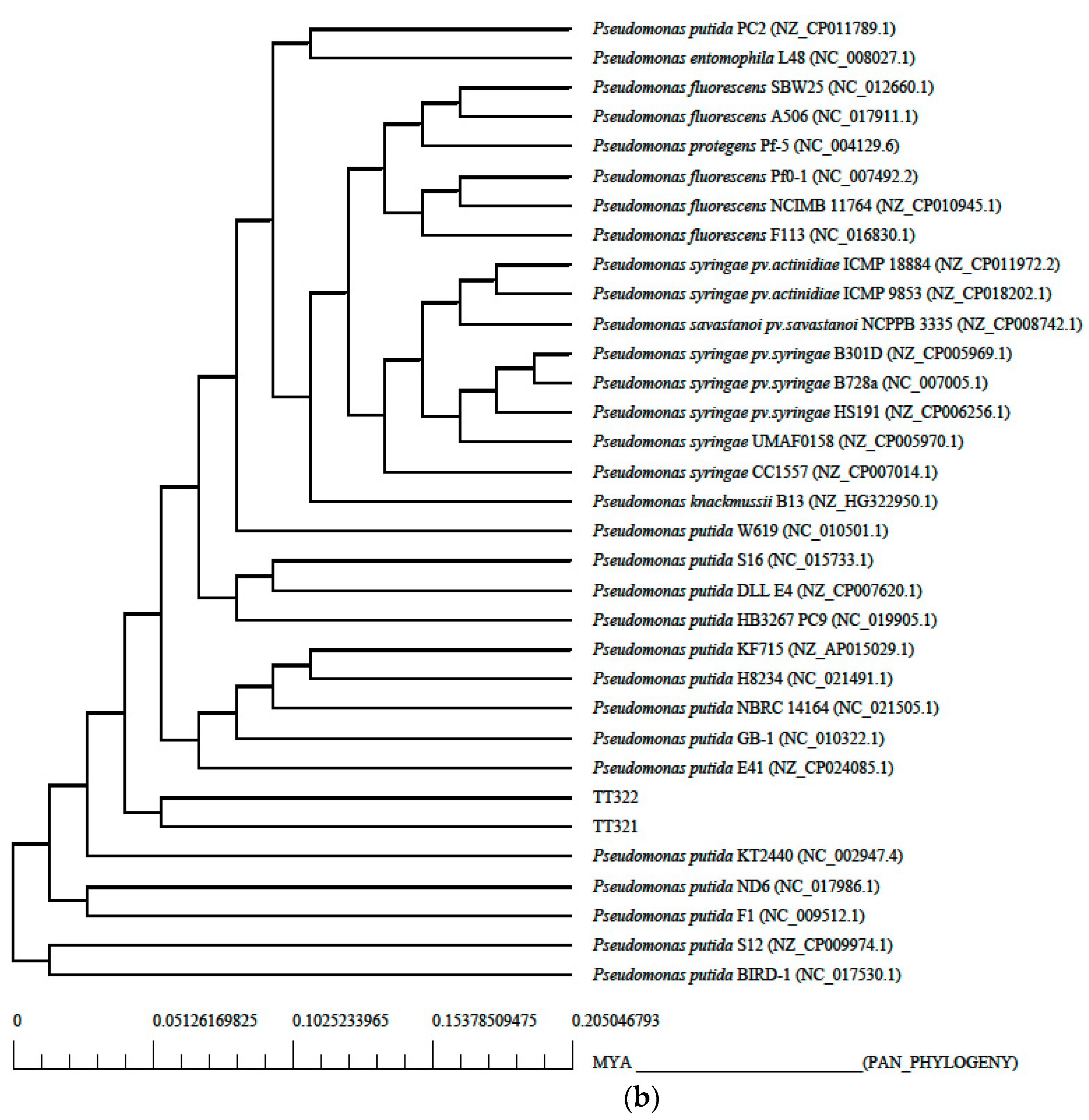
3.2. Phylogenetic Tree Analysis
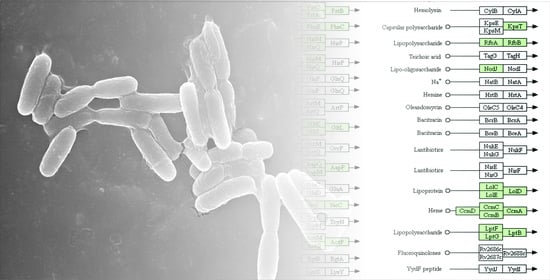
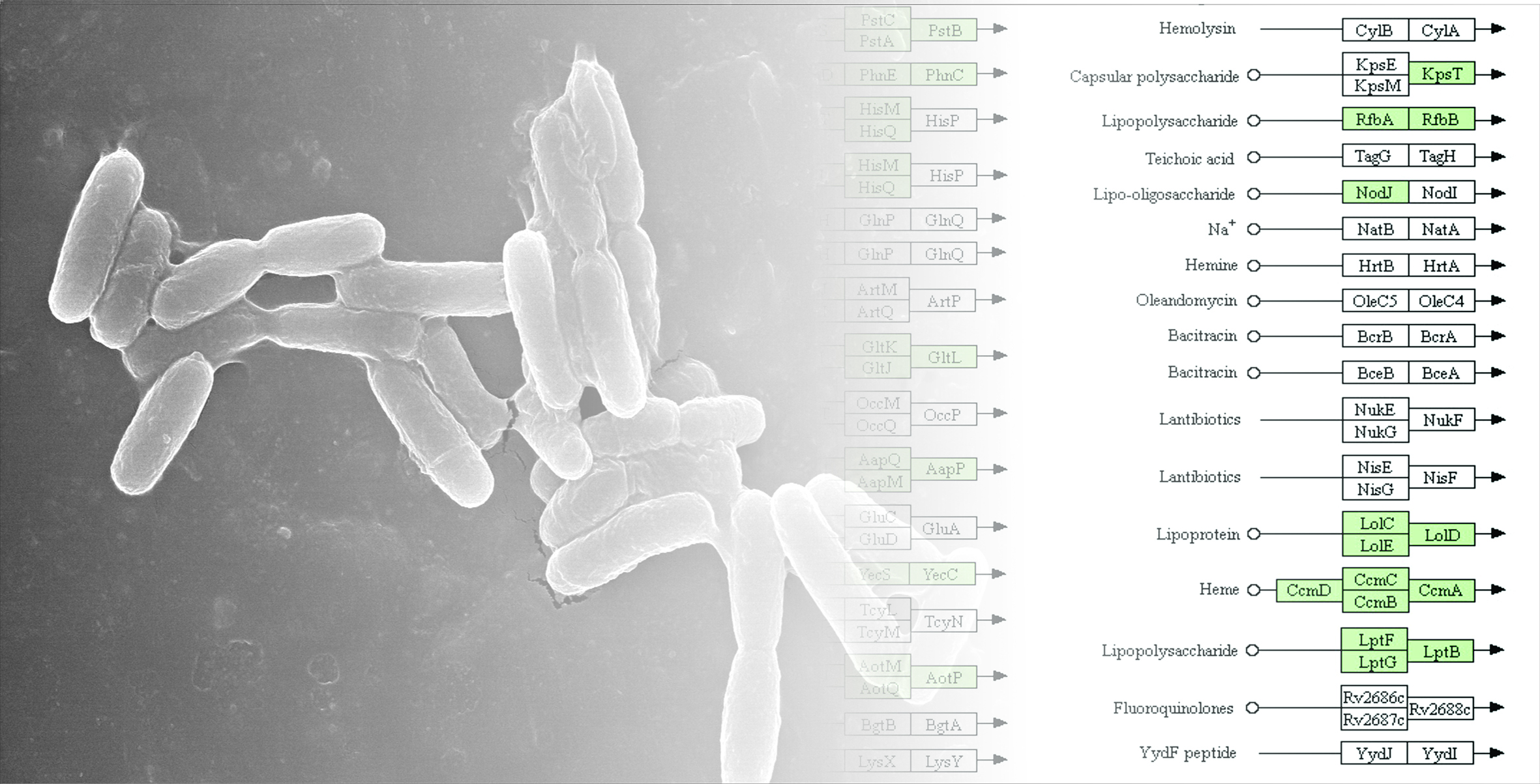
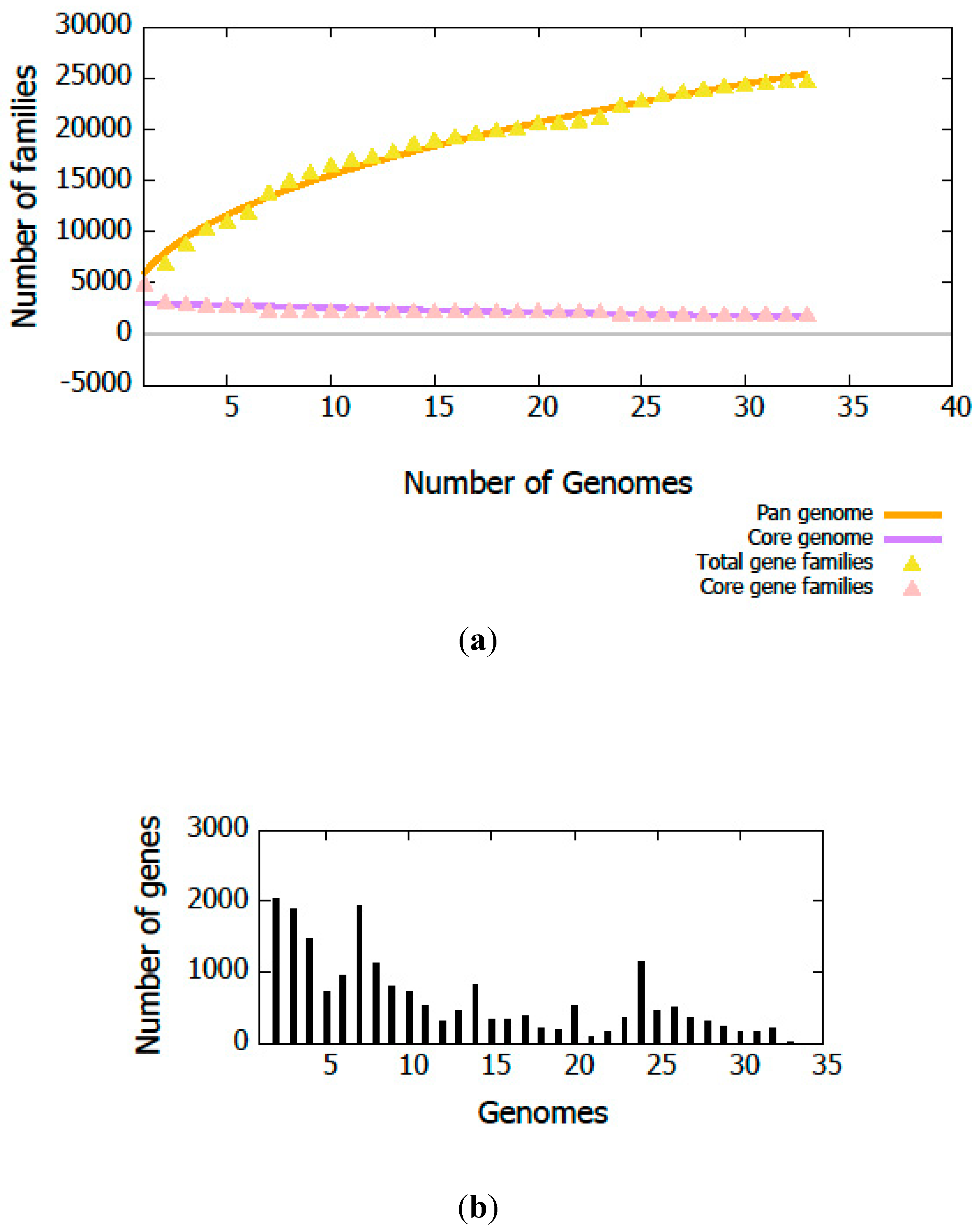
3.3. Genome Annotation and Gene Repertoire
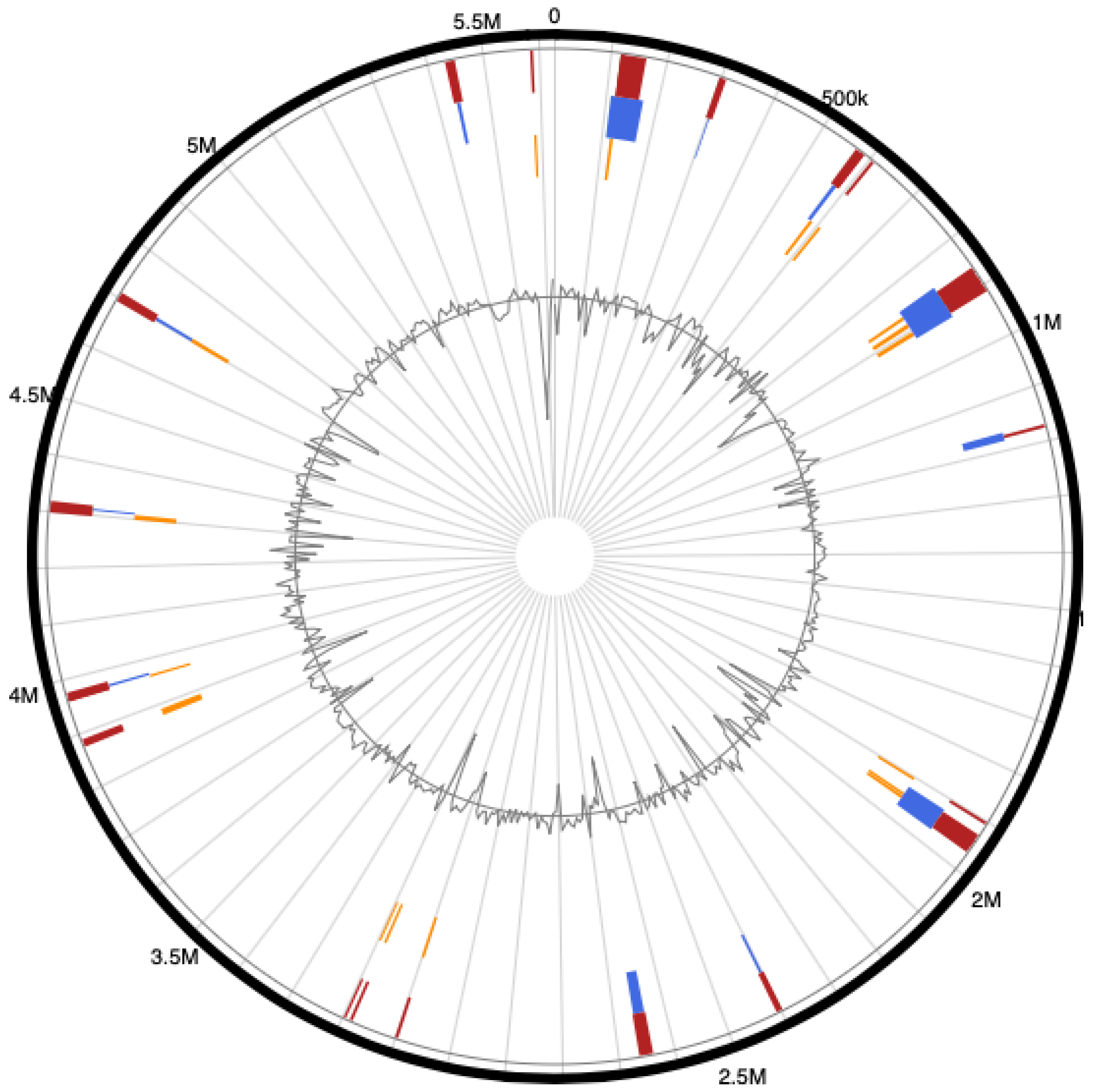
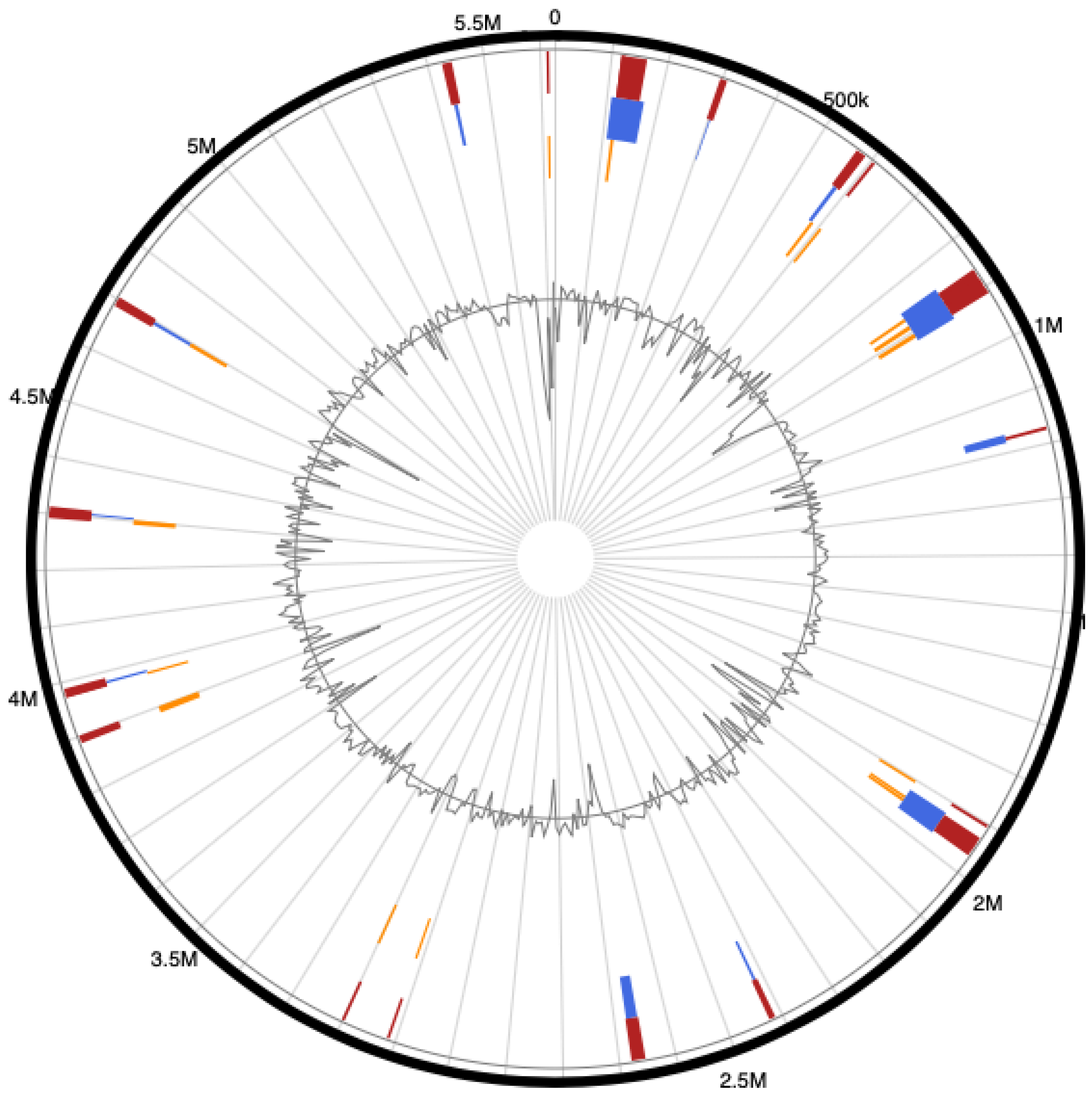
3.4. Genome Islands (GIs) of Pseudomonas
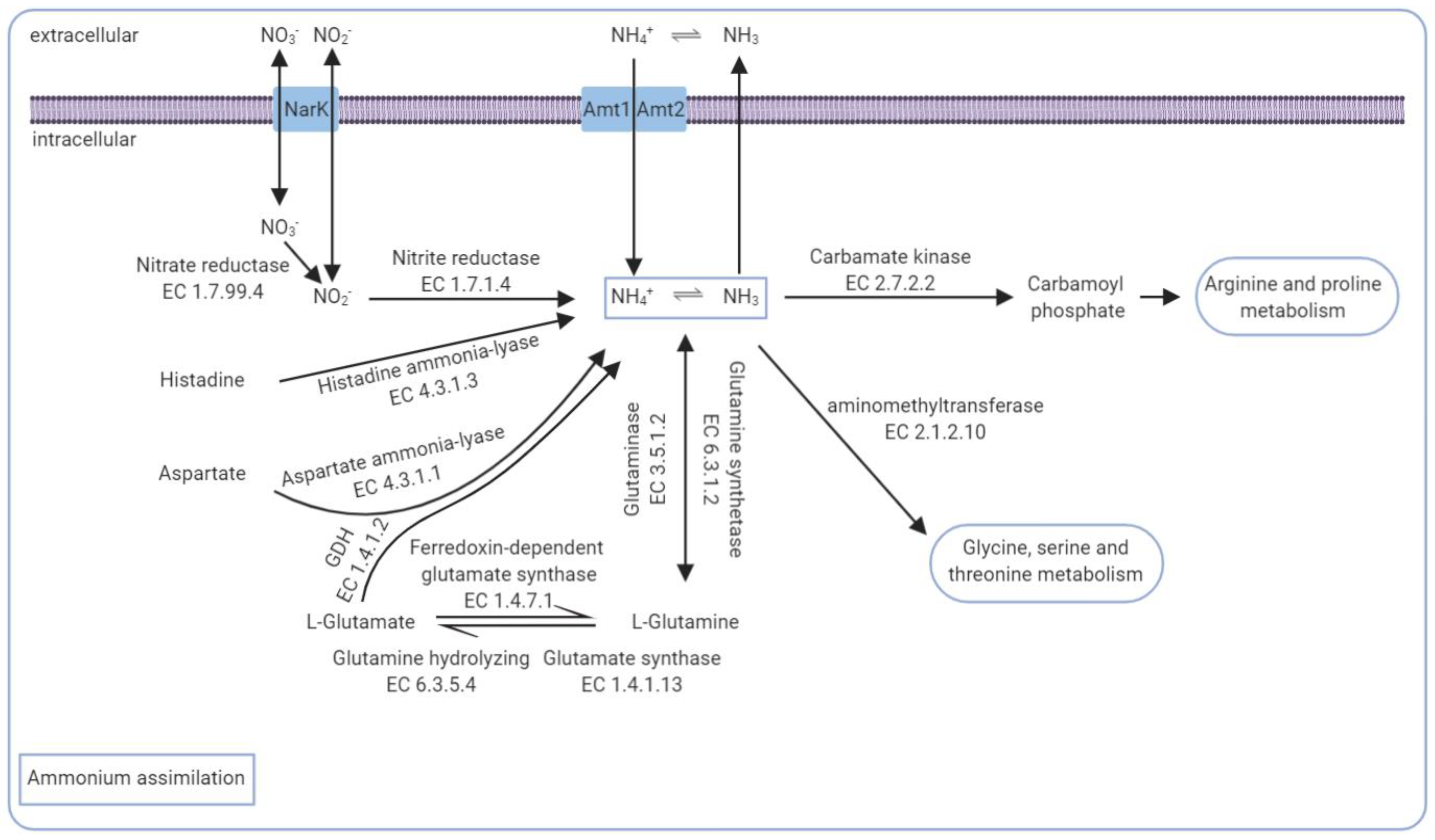
3.5. The Core Genomic Repertoire Involved in Ammonia Conversion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sah, S.; Krishnani, S.; Singh, R. Pseudomonas Mediated Nutritional and Growth Promotional Activities for Sustainable Food Security. Curr. Res. Microb. Sci. 2021, 2, 100084. [Google Scholar] [CrossRef]
- Gomila, M.; Peña, A.; Mulet, M.; Lalucat, J.; García-Valdés, E. Phylogenomics and Systematics in Pseudomonas. Front. Microbiol. 2015, 6, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, E.R.B.; Tindall, B.J.; Martins Dos Santos, V.A.P.; Pieper, D.H.; Ramos, J.-L.; Palleroni, N.J. Nonmedical: Pseudomonas. In The Prokaryotes: Volume 6: Proteobacteria: Gamma Subclass; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 646–703. ISBN 978-0-387-30746-6. [Google Scholar]
- Bardera, G.; Usman, N.; Owen, M.; Pountney, D.; Sloman, K.A.; Alexander, M.E. The Importance of Behaviour in Improving the Production of Shrimp in Aquaculture. Rev. Aquac. 2019, 11, 1104–1132. [Google Scholar] [CrossRef]
- Jackson, C.; Preston, N.; Thompson, P.J.; Burford, M. Nitrogen Budget and Effluent Nitrogen Components at an Intensive Shrimp Farm. Aquaculture 2003, 218, 397–411. [Google Scholar] [CrossRef]
- Burford, M.A.; Lorenzen, K. Modeling Nitrogen Dynamics in Intensive Shrimp Ponds: The Role of Sediment Remineralization. Aquaculture 2004, 229, 129–145. [Google Scholar] [CrossRef] [Green Version]
- Kuypers, M.M.M.; Marchant, H.K.; Kartal, B. The Microbial Nitrogen-Cycling Network. Nat. Rev. Microbiol. 2018, 16, 263–276. [Google Scholar] [CrossRef]
- Chang, H.; Yang, X.; Fang, Y.; Pu, P.; Li, Z.; Rengel, Z. In-Situ Nitrogen Removal from the Eutrophic Water by Microbial-Plant Integrated System. J. Zhejiang Univ. Sci. B 2006, 7, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.M.; He, Y.; Zhao, B.; Huang, J. Heterotrophic Ammonium Removal Characteristics of an Aerobic Heterotrophic Nitrifying-Denitrifying Bacterium, Providencia Rettgeri YL. J. Environ. Sci. 2009, 21, 1336–1341. [Google Scholar] [CrossRef]
- Zhang, Q.-L.; Liu, Y.; Ai, G.-M.; Miao, L.-L.; Zheng, H.-Y.; Liu, Z.-P. The Characteristics of a Novel Heterotrophic Nitrification–Aerobic Denitrification Bacterium, Bacillus Methylotrophicus Strain L7. Bioresour. Technol. 2012, 108, 35–44. [Google Scholar] [CrossRef]
- He, T.; Li, Z.; Sun, Q.; Xu, Y.; Ye, Q. Heterotrophic Nitrification and Aerobic Denitrification by Pseudomonas Tolaasii Y-11 without Nitrite Accumulation during Nitrogen Conversion. Bioresour. Technol. 2016, 200, 493–499. [Google Scholar] [CrossRef]
- Jin, R.; Liu, T.; Liu, G.; Zhou, J.; Huang, J.; Wang, A. Simultaneous Heterotrophic Nitrification and Aerobic Denitrification by the Marine Origin Bacterium pseudomonas sp. ADN-42. Appl. Biochem. Biotechnol. 2015, 175, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; He, T.; Li, Z.; Ye, Q.; Chen, Y.; Xie, E.; Zhang, X. Nitrogen Removal Characteristics of Pseudomonas Putida Y-9 Capable of Heterotrophic Nitrification and Aerobic Denitrification at Low Temperature. Available online: https://www.hindawi.com/journals/bmri/2017/1429018/ (accessed on 13 October 2017).
- Trung Tran, T.; Bott, N.J.; Dai Lam, N.; Trung Nguyen, N.; Hoang Thi Dang, O.; Hoang Le, D.; Tung Le, L.; Hoang Chu, H. The Role of Pseudomonas in Heterotrophic Nitrification: A Case Study on Shrimp Ponds (Litopenaeus vannamei) in Soc Trang Province. Microorganisms 2019, 7, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an Ultra-Fast Pan-Genome Analysis Pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded Prediction of Genomic Islands for Larger-Scale Datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Cordero, O.X.; Hogeweg, P. The Impact of Long-Distance Horizontal Gene Transfer on Prokaryotic Genome Size. Proc. Natl. Acad. Sci. USA 2009, 106, 21748–21753. [Google Scholar] [CrossRef] [Green Version]
- Fuchsman, C.A.; Collins, R.E.; Rocap, G.; Brazelton, W.J. Effect of the Environment on Horizontal Gene Transfer between Bacteria and Archaea. PeerJ 2017, 5, e3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gophna, U.; Kristensen, D.M.; Wolf, Y.I.; Popa, O.; Drevet, C.; Koonin, E.V. No Evidence of Inhibition of Horizontal Gene Transfer by CRISPR-Cas on Evolutionary Timescales. ISME J. 2015, 9, 2021–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Church, D.L.; Cerutti, L.; Gürtler, A.; Griener, T.; Zelazny, A.; Emler, S. Performance and Application of 16S RRNA Gene Cycle Sequencing for Routine Identification of Bacteria in the Clinical Microbiology Laboratory. Clin. Microbiol. Rev. 2020, 33, e00053-19. [Google Scholar] [CrossRef] [PubMed]
- Kerou, M.; Offre, P.; Valledor, L.; Abby, S.S.; Melcher, M.; Nagler, M.; Weckwerth, W.; Schleper, C. Proteomics and Comparative Genomics of Nitrososphaera Viennensis Reveal the Core Genome and Adaptations of Archaeal Ammonia Oxidizers. Proc. Natl. Acad. Sci. USA 2016, 113, E7937–E7946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmis, K.N. Pseudomonas Putida: A Cosmopolitan Opportunist Par Excellence. Environ. Microbiol. 2002, 4, 779–781. [Google Scholar] [CrossRef]
- Miyazaki, R.; Bertelli, C.; Benaglio, P.; Canton, J.; De Coi, N.; Gharib, W.H.; Gjoksi, B.; Goesmann, A.; Greub, G.; Harshman, K.; et al. Comparative Genome Analysis of Pseudomonas Knackmussii B13, the First Bacterium Known to Degrade Chloroaromatic Compounds. Environ. Microbiol. 2015, 17, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Blanco, A.; Blanco, G. Chapter 16—Amino Acid Metabolism. In Medical Biochemistry; Blanco, A., Blanco, G., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 367–399. ISBN 978-0-12-803550-4. [Google Scholar]
- Okamura-Ikeda, K.; Hosaka, H.; Maita, N.; Fujiwara, K.; Yoshizawa, A.C.; Nakagawa, A.; Taniguchi, H. Crystal Structure of Aminomethyltransferase in Complex with Dihydrolipoyl-H-Protein of the Glycine Cleavage System: Implications for Recognition of Lipoyl Protein Substrate, Disease-Related Mutations, and Reaction Mechanism. J. Biol. Chem. 2010, 285, 18684–18692. [Google Scholar] [CrossRef] [Green Version]
- Moir, J.W.; Wood, N.J. Nitrate and Nitrite Transport in Bacteria. Cell. Mol. Life Sci. 2001, 58, 215–224. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession a | Organism | Size (bp) | GC b (%) | CDSs c | No. Plasmids d | No. Contigs e | Subsystems f |
|---|---|---|---|---|---|---|---|
| This study | Pseudomonas putida TT321 | 5,566,241 | 61.8 | 5210 | 0 | 62 | 393 |
| This study | Pseudomonas putida TT322 | 5,563,644 | 61.8 | 5234 | 0 | 64 | 393 |
| NC_016830.1 | Pseudomonas fluorescens F113 | 6,845,832 | 60.8 | 6249 | 0 | 1 | 410 |
| NC_012660.1 | Pseudomonas fluorescens SBW25 | 6,722,539 | 60.5 | 6162 | 0 | 1 | 413 |
| NC_017530.1 | Pseudomonas putida BIRD-1 | 5,731,541 | 61.7 | 5297 | 0 | 1 | 393 |
| NC_009512.1 | Pseudomonas putida F1 | 5,959,964 | 61.9 | 5436 | 0 | 1 | 395 |
| NC_010322.1 | Pseudomonas putida GB-1 | 6,078,430 | 61.9 | 5541 | 0 | 1 | 397 |
| NC_021491.1 | Pseudomonas putida H8234 | 6,870,827 | 61.6 | 6593 | 0 | 1 | 409 |
| NZ_CP007620.1 | Pseudomonas putida DLL-E4 | 6,484,062 | 62.5 | 6252 | 0 | 1 | 399 |
| NC_002947.4 | Pseudomonas putida KT2440 | 6,181,873 | 61.5 | 5716 | 0 | 1 | 402 |
| NC_021505.1 | Pseudomonas putida NBRC 14164 | 6,156,701 | 62.3 | 5556 | 0 | 1 | 402 |
| NZ_CP011789.1 | Pseudomonas putida PC2 | 5,808,624 | 63.2 | 5232 | 0 | 1 | 390 |
| NC_015733.1 | Pseudomonas putida S16 | 5,984,790 | 62.3 | 5701 | 0 | 1 | 404 |
| NC_010501.1 | Pseudomonas putida W619 | 5,774,330 | 61.4 | 5357 | 0 | 1 | 405 |
| NZ_CP005969.1 | Pseudomonas syringae pv. syringae B301D | 6,094,819 | 59.2 | 5413 | 0 | 1 | 389 |
| NC_007005.1 | Pseudomonas syringae pv. syringae B728a | 6,093,698 | 59.2 | 5395 | 0 | 1 | 389 |
| NC_008027.1 | Pseudomonas entomophila L48 | 5,888,780 | 64.2 | 5209 | 0 | 1 | 397 |
| NC_017911.1 | Pseudomonas fluorescens A506 | 5,962,570 | 60.0 | 5505 | 1 | 1 | 400 |
| NZ_CP010945.1 | Pseudomonas fluorescens NCIMB 11764 | 6,998,154 | 59.0 | 6564 | 0 | 1 | 413 |
| NC_007492.2 | Pseudomonas fluorescens Pf0-1 | 6,438,405 | 60.5 | 5840 | 0 | 1 | 410 |
| NZ_HG322950.1 | Pseudomonas knackmussii B13 | 6,162,905 | 65.6 | 5887 | 0 | 1 | 387 |
| NC_004129.6 | Pseudomonas protegens Pf-5 | 7,074,893 | 63.3 | 6412 | 0 | 1 | 416 |
| NZ_CP024085.1 | Pseudomonas putida E41 | 6,093,023 | 62.2 | 5433 | 0 | 1 | 407 |
| NC_019905.1 | Pseudomonas putida HB3267 | 5,875,750 | 62.6 | 5475 | 1 | 1 | 401 |
| NZ_AP015029.1 | Pseudomonas putida KF715 | 6,583,377 | 61.9 | 6072 | 7 | 1 | 419 |
| NC_017986.1 | Pseudomonas putida ND6 | 6,085,449 | 61.8 | 5632 | 2 | 1 | 401 |
| NZ_CP009974.1 | Pseudomonas putida S12 | 5,798,534 | 61.8 | 5269 | 1 | 1 | 394 |
| NZ_CP008742.1 | Pseudomonas savastanoi pv. savastanoi NCPPB 3335 | 6,016,828 | 58.1 | 5800 | 0 | 1 | 383 |
| NZ_CP007014.1 | Pseudomonas syringae CC1557 | 5,758,024 | 58.6 | 5244 | 0 | 1 | 378 |
| NZ_CP018202.1 | Pseudomonas syringae pv. actinidiae ICMP 9853 | 6,439,609 | 58.7 | 6046 | 0 | 1 | 390 |
| NZ_CP011972.2 | Pseudomonas syringae pv. actinidiae ICMP 18884 | 6,555,569 | 58.4 | 6183 | 1 | 1 | 387 |
| NZ_CP006256.1 | Pseudomonas syringae pv. syringae HS191 | 5,950,211 | 59.0 | 5271 | 1 | 1 | 389 |
| NZ_CP005970.1 | Pseudomonas syringae UMAF0158 | 5,787,986 | 59.3 | 5145 | 1 | 1 | 384 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, T.T.; Bott, N.J.; Gelderen, R.v.; Nguyen, N.T.; Chu, P.M.T.; Chu, H.H. Comparative Genome Analysis of Two Heterotrophic Nitrifying Pseudomonas putida Strains Isolated from Freshwater Shrimp Ponds in Soc Trang Province. Fermentation 2022, 8, 336. https://doi.org/10.3390/fermentation8070336
Tran TT, Bott NJ, Gelderen Rv, Nguyen NT, Chu PMT, Chu HH. Comparative Genome Analysis of Two Heterotrophic Nitrifying Pseudomonas putida Strains Isolated from Freshwater Shrimp Ponds in Soc Trang Province. Fermentation. 2022; 8(7):336. https://doi.org/10.3390/fermentation8070336
Chicago/Turabian StyleTran, Thanh Trung, Nathan J. Bott, Rebecca van Gelderen, Nam Trung Nguyen, Phuong Minh Thi Chu, and Ha Hoang Chu. 2022. "Comparative Genome Analysis of Two Heterotrophic Nitrifying Pseudomonas putida Strains Isolated from Freshwater Shrimp Ponds in Soc Trang Province" Fermentation 8, no. 7: 336. https://doi.org/10.3390/fermentation8070336