Carbons as Catalysts in Thermo-Catalytic Hydrocarbon Decomposition: A Review

Abstract
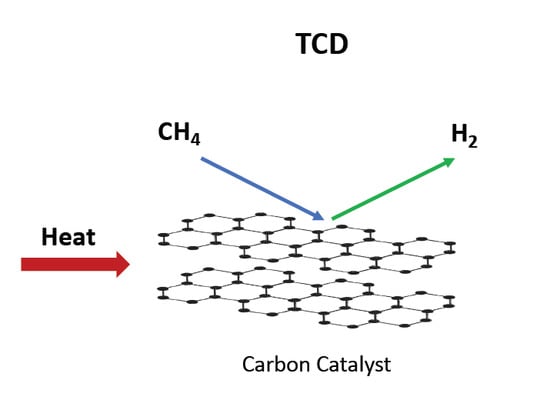
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Sustainability of Thermo-Catalytic Decomposition of Methane and Natural Gas Components
- Decarbonizing a fossil fuel for clean H2, providing a bridge to the H2 economy;
- Enabling renewable energy coupling/storage into chemical bond energy;
- Producing solid carbons as energy storage media, for potential structural materials and realizing at-scale solid carbon sequestration.
2. Thermo-Catalytic (Methane) Decomposition—Literature Analysis
2.1. Deposition
2.2. Continued Studies Using Porous or Activated Carbons
2.3. Novel Studies
2.4. Electron Microscopy in TCD
2.5. Results with Other Hydrocarbon Gases
2.6. CO2 Gasification of TCD Carbon
- Gasification reactivity increased for higher TCD (decomposition) temperatures—rationalized by smaller crystallites (and hence more edge sites) being produced by the faster (and presumably less ordered) deposition at the higher temperatures. (This indicates that structure of deposit depends on process parameters). Moreover, this implies that temperature affects the deposition process but not the carbon once deposited.
- With increasing (gasification) conversion, i.e., burnoff, activation energies decreased. (This suggests variation in deposit structure, with less order for the innermost portion—the earliest deposition). Notably, this is the reciprocal observation of the decline in rate with increasing deposition in nearly all studies—where the later deposit has the lowest activity and rate (attributed to higher order). That highlights the complementarity of gasification for revealing the structure of the deposited carbon. In most studies only the final deactivated catalyst is characterized, not intermediate stages. The proposed HRTEM of the deposit/catalyst at intermediate stages will address this. Atomistic simulations will co-address both deposit nanostructure evolution and its temperature dependence.
2.7. Conceptualization of Carbon Deposition in TCD and Removal via Regeneration
2.8. Economic Analyses of TCD
3. Addressing TCD Research Needs
4. TCD Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abbas, H.F.; Daud, W.W. Hydrogen production by methane decomposition: A review. Int. J. Hydrogen Energy 2010, 35, 1160–1190. [Google Scholar] [CrossRef]
- Marbán, G.; Valdés-Solís, T. Towards the hydrogen economy? Int. J. Hydrogen Energy 2007, 32, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Spath, P.L.; Mann, M.K. Life Cycle Assessment of Hydrogen Production via Natural Gas Steam Reforming; Technical Report NREL/TP-570-27637; National Renewable Energy Lab: Golden, CO, USA, February 2001. Available online: https://www.osti.gov/biblio/764485 (accessed on 10 April 2020).
- Muradov, N. Hydrogen via methane decomposition: An application for decarbonization of fossil fuels. Int. J. Hydrogen Energy 2001, 26, 1165–1175. [Google Scholar] [CrossRef]
- Muradov, N. Thermocatalytic CO2-free production of hydrogen from hydrocarbon fuels. In Proceedings of the 2000 Hydrogen Program Review, NREL/CP-570-28890; May 2000. Available online: https://www.nrel.gov/docs/fy01osti/28890.pdf (accessed on 10 April 2020).
- Ahmed, S.; Aitani, A.; Rahman, F.; Al-Dawood, A.; Al-Muhaish, F. Decomposition of hydrocarbons to hydrogen and carbon. Appl. Catal. A Gen. 2009, 359, 1–24. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar thermochemical production of hydrogen—A review. Sol. Energy 2005, 78, 603–615. [Google Scholar] [CrossRef]
- Abanades, S.; Flamant, G. Experimental study and modeling of a high-temperature solar chemical reactor for hydrogen production from methane cracking. Int. J. Hydrogen Energy 2007, 32, 1508–1515. [Google Scholar] [CrossRef]
- Amin, A.M.; Croiset, E.; Epling, W. Review of methane catalytic cracking for hydrogen production. Int. J. Hydrogen Energy 2011, 36, 2904–2935. [Google Scholar] [CrossRef]
- Dufour, J.; Serrano, D.P.; Galvez, J.L.; Moreno, J.; Garcia, C. Life cycle assessment of processes for hydrogen production. Environmental feasibility and reduction of greenhouse gases emissions. Int. J. Hydrogen Energy 2009, 34, 1370–1376. [Google Scholar] [CrossRef]
- Lane, J.M.; Spath, P.L. Technoeconomic Analysis of the Thermocatalytic Decomposition of Natural Gas; Department of Energy: Golden, CO, USA, 2001.
- Steinberg, M. Fossil fuel decarbonization technology for mitigating global warming. Int. J. Hydrogen Energy 1999, 24, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Ashik, U.P.M.; Daud, W.W.; Abbas, H.F. Production of greenhouse gas free hydrogen by thermocatalytic decomposition of methane–a review. Renew. Sustain. Energy Rev. 2015, 44, 221–256. [Google Scholar] [CrossRef] [Green Version]
- Gaudernack, B.; Lynum, S. Hydrogen from natural gas without release of CO2 to the atmosphere. Int. J. Hydrogen Energy 1998, 23, 1087–1093. [Google Scholar] [CrossRef]
- Srilatha, K.; Viditha, V.; Srinivasulu, D.; Ramakrishna, S.U.B.; Himabindu, V. Hydrogen production using thermocatalytic decomposition of methane on Ni30/activated carbon and Ni30/carbon black. Environ. Sci. Pollut. Res. 2016, 23, 9303–9311. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Croiset, E.; Hudgins, R.R. Catalytic decomposition of methane for hydrogen production. Top. Catal. 2006, 37, 137–145. [Google Scholar] [CrossRef]
- De Falco, M.; Basile, A. Enriched Methane: The First Step Towards the Hydrogen Economy; Springer: Switzerland, 2015. [Google Scholar]
- Aiello, R.; Fiscus, J.E.; zur Loye, H.C.; Amiridis, M.D. Hydrogen production via the direct cracking of methane over Ni/SiO2: Catalyst deactivation and regeneration. Appl. Catal. A Gen. 2000, 192, 227–234. [Google Scholar] [CrossRef]
- Bai, Z.; Chen, H.; Li, B.; Li, W. Catalytic decomposition of methane over activated carbon. J. Anal. Appl. Pyrolysis 2005, 73, 335–341. [Google Scholar] [CrossRef]
- Lee, K.K.; Han, G.Y.; Yoon, K.J.; Lee, B.K. Thermocatalytic hydrogen production from the methane in a fluidized bed with activated carbon catalyst. Catal. Today 2004, 93, 81–86. [Google Scholar] [CrossRef]
- Muradov, N.; Smith, F.; Ali, T. Catalytic activity of carbons for methane decomposition reaction. Catal. Today 2005, 102, 225–233. [Google Scholar] [CrossRef]
- Jung, J.U.; Nam, W.; Yoon, K.J.; Han, G.Y. Hydrogen production by catalytic decomposition of methane over carbon catalysts in a fluidized bed. Korean J. Chem. Eng. 2007, 24, 674–678. [Google Scholar] [CrossRef]
- Dunker, A.M.; Kumar, S.; Mulawa, P.A. Production of hydrogen by thermal decomposition of methane in a fluidized-bed reactor—Effects of catalyst, temperature, and residence time. Int. J. Hydrogen Energy 2006, 31, 473–484. [Google Scholar] [CrossRef]
- Abbas, H.F.; Daud, W.W. Hydrogen production by thermocatalytic decomposition of methane using a fixed bed activated carbon in a pilot scale unit: Apparent kinetic, deactivation and diffusional limitation studies. Int. J. Hydrogen Energy 2010, 35, 12268–12276. [Google Scholar] [CrossRef]
- Abbas, H.F.; Daud, W.W. Thermocatalytic decomposition of methane using palm shell based activated carbon: Kinetic and deactivation studies. Fuel Process. Technol. 2009, 90, 1167–1174. [Google Scholar] [CrossRef]
- Kim, M.H.; Lee, E.K.; Jun, J.H.; Han, G.Y.; Kong, S.J.; Lee, B.K.; Yoon, K.J. Hydrogen production by catalytic decomposition of methane over activated carbons: Deactivation study. Korean J. Chem. Eng. 2003, 20, 835–839. [Google Scholar] [CrossRef]
- Kim, M.H.; Lee, E.K.; Jun, J.H.; Kong, S.J.; Han, G.Y.; Lee, B.K.; Yoon, K.J. Hydrogen production by catalytic decomposition of methane over activated carbons: Kinetic study. Int. J. Hydrogen Energy 2004, 29, 187–193. [Google Scholar] [CrossRef]
- Moliner, R.; Suelves, I.; Lázaro, M.J.; Moreno, O. Thermocatalytic decomposition of methane over activated carbons: Influence of textural properties and surface chemistry. Int. J. Hydrogen Energy 2005, 30, 293–300. [Google Scholar] [CrossRef]
- Ashok, J.; Kumar, S.N.; Venugopal, A.; Kumari, V.D.; Tripathi, S.; Subrahmanyam, M. COx free hydrogen by methane decomposition over activated carbons. Catal. Commun. 2008, 9, 164–169. [Google Scholar] [CrossRef]
- Lee, E.K.; Lee, S.Y.; Han, G.Y.; Lee, B.K.; Lee, T.J.; Jun, J.H.; Yoon, K.J. Catalytic decomposition of methane over carbon blacks for CO 2-free hydrogen production. Carbon 2004, 42, 2641–2648. [Google Scholar] [CrossRef]
- Ryu, B.H.; Lee, S.Y.; Lee, D.H.; Han, G.Y.; Lee, T.J.; Yoon, K.J. Catalytic characteristics of various rubber-reinforcing carbon blacks in decomposition of methane for hydrogen production. Catal. Today 2007, 123, 303–309. [Google Scholar] [CrossRef]
- Suelves, I.; Lázaro, M.J.; Moliner, R.; Pinilla, J.L.; Cubero, H. Hydrogen production by methane decarbonization: Carbonaceous catalysts. Int. J. Hydrogen Energy 2007, 32, 3320–3326. [Google Scholar] [CrossRef]
- Pinilla, J.L.; Suelves, I.; Lázaro, M.J.; Moliner, R. Kinetic study of the thermal decomposition of methane using carbonaceous catalysts. Chem. Eng. J. 2008, 138, 301–306. [Google Scholar] [CrossRef]
- Suelves, I.; Pinilla, J.L.; Lázaro, M.J.; Moliner, R. Carbonaceous materials as catalysts for decomposition of methane. Chem. Eng. J. 2008, 140, 432–438. [Google Scholar] [CrossRef]
- Bai, Z.; Chen, H.; Li, W.; Li, B. Hydrogen production by methane decomposition over coal char. Int. J. Hydrogen Energy 2006, 31, 899–905. [Google Scholar] [CrossRef]
- Sun, Z.Q.; Wu, J.H.; Haghighi, M.; Bromly, J.; Ng, E.; Wee, H.L.; Zhang, D.K. Methane cracking over a bituminous coal char. Energy Fuels 2007, 21, 1601–1605. [Google Scholar] [CrossRef]
- Lázaro, M.J.; Pinilla, J.L.; Suelves, I.; Moliner, R. Study of the deactivation mechanism of carbon blacks used in methane decomposition. Int. J. Hydrogen Energy 2008, 33, 4104–4111. [Google Scholar] [CrossRef]
- Muradov, N. Thermocatalytic CO2-Free Production of Hydrogen from Hydrocarbon Fuels; U.S. DOE Hydrogen Program Review. U.S.: Department of Energy (DOE); 2002; NREL/CP-610-32405. Available online: https://www.nrel.gov/docs/fy02osti/32405a17.pdf (accessed on 10 April 2020).
- Bai, Z.; Li, W.; Bai, J.; Li, B.; Chen, H. The effects of textural properties and surface chemistry of activated carbon on its catalytic performance in methane decomposition for hydrogen production. Energy Sources Part A Recovery Util. Environ. Eff. 2012, 34, 1145–1153. [Google Scholar] [CrossRef]
- Pinilla, J.L.; Suelves, I.; Lázaro, M.J.; Moliner, R. Influence on hydrogen production of the minor components of natural gas during its decomposition using carbonaceous catalysts. J. Power Sources 2009, 192, 100–106. [Google Scholar] [CrossRef]
- Muradov, N.; Smith, F.; Bokerman, G. Methane activation by nonthermal plasma generated carbon aerosols. J. Phys. Chem. C 2009, 113, 9737–9747. [Google Scholar] [CrossRef]
- Moliner, R.; Suelves, I.; Lazaro, M.J.; Corbella, B.M.; Palacios, J.M. Hydrogen production by catalytic decomposition of methane over carbonaceous materials. In Proceedings of the International Conference on Carbon, Oviedo, Spain, 6–10 July 2003. [Google Scholar]
- Pinilla, J.L.; Suelves, I.; Utrilla, R.; Gálvez, M.E.; Lázaro, M.J.; Moliner, R. Hydrogen production by thermo-catalytic decomposition of methane: Regeneration of active carbons using CO2. J. Power Sources 2007, 169, 103–109. [Google Scholar] [CrossRef]
- Serrano, D.P.; Botas, J.A.; Guil-Lopez, R. H2 production from methane pyrolysis over commercial carbon catalysts: Kinetic and deactivation study. Int. J. Hydrogen Energy 2009, 34, 4488–4494. [Google Scholar] [CrossRef]
- Serrano, D.P.; Botas, J.A.; Fierro, J.L.G.; Guil-López, R.; Pizarro, P.; Gómez, G. Hydrogen production by methane decomposition: Origin of the catalytic activity of carbon materials. Fuel 2010, 89, 1241–1248. [Google Scholar] [CrossRef]
- Krzyżyński, S.; Kozłowski, M. Activated carbons as catalysts for hydrogen production via methane decomposition. Int. J. Hydrogen Energy 2008, 33, 6172–6177. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ryu, B.H.; Han, G.Y.; Lee, T.J.; Yoon, K.J. Catalytic characteristics of specialty carbon blacks in decomposition of methane for hydrogen production. Carbon 2008, 46, 1978–1986. [Google Scholar] [CrossRef]
- Malaika, A.; Kozłowski, M. Influence of ethylene on carbon-catalysed decomposition of methane. Int. J. Hydrogen Energy 2009, 34, 2600–2605. [Google Scholar] [CrossRef]
- Kameya, Y.; Hanamura, K. Carbon black texture evolution during catalytic methane decomposition. Carbon 2012, 50, 3503–3512. [Google Scholar] [CrossRef]
- Müller, J.O.; Su, D.S.; Jentoft, R.E.; Kröhnert, J.; Jentoft, F.C.; Schlögl, R. Morphology-controlled reactivity of carbonaceous materials towards oxidation. Catal. Today 2005, 102, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Alam, M.; Boehman, A.L.; Kim, U. Examination of the oxidation behavior of biodiesel soot. Combust. Flame 2006, 146, 589–604. [Google Scholar] [CrossRef]
- Al-Qurashi, K.; Boehman, A.L. Impact of exhaust gas recirculation (EGR) on the oxidative reactivity of diesel engine soot. Combust. Flame 2008, 155, 675–695. [Google Scholar] [CrossRef]
- Knauer, M.; Schuster, M.E.; Su, D.; Schlögl, R.; Niessner, R.; Ivleva, N.P. Soot structure and reactivity analysis by Raman microspectroscopy, temperature-programmed oxidation, and high-resolution transmission electron microscopy. J. Phys. Chem. A 2009, 113, 13871–13880. [Google Scholar] [CrossRef]
- Liu, F.; Yang, L.; Song, C. Chemical looping hydrogen production using activated carbon and carbon black as multi-function carriers. Int. J. Hydrogen Energy 2018, 43, 5501–5511. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Dentzer, J.; Gadiou, R.; Ouederni, A. Evaluation of activated carbons based on olive stones as catalysts during hydrogen production by thermocatalytic decomposition of methane. Int. J. Hydrogen Energy 2017, 42, 8712–8720. [Google Scholar] [CrossRef]
- Wang, J.; Jin, L.; Zhou, Y.; Li, Y.; Hu, H. Effect of Ca (NO3) 2 addition in coal on properties of activated carbon for methane decomposition to hydrogen. Fuel Process. Technol. 2018, 176, 85–90. [Google Scholar] [CrossRef]
- Nishii, H.; Miyamoto, D.; Umeda, Y.; Hamaguchi, H.; Suzuki, M.; Tanimoto, T.; Harigai, T.; Takikawa, H.; Suda, Y. Catalytic activity of several carbons with different structures for methane decomposition and by-produced carbons. Appl. Surf. Sci. 2019, 473, 291–297. [Google Scholar] [CrossRef]
- Więckowski, A.B.; Najder-Kozdrowska, L.; Rechnia, P.; Malaika, A.; Krzyżyńska, B.; Kozłowski, M. EPR Characteristics of Activated Carbon for Hydrogen Production by the Thermo-Catalytic Decomposition of Methane. Acta Phys. Pol. A 2016, 130, 701–704. [Google Scholar] [CrossRef]
- Ghani, S.A.; Al Saraj, M.A.A.; Razaq, G.A. Catalytic Production of COx Free Hydrogen by Methane Decomposition over Activated Carbons. Energy Sources Part A Recovery Util. Environ. Eff. 2015, 37, 326–333. [Google Scholar] [CrossRef]
- Shen, Y.; Lua, A.C. A trimodal porous carbon as an effective catalyst for hydrogen production by methane decomposition. J. Colloid Interface Sci. 2016, 462, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Shilapuram, V.; Ozalp, N. Hydrogen production by carbon-catalyzed methane decomposition via thermogravimetry. J. Energy Resour. Technol. 2017, 139, 012005. [Google Scholar] [CrossRef]
- Luo, H.; Qiao, Y.; Ning, Z.; Bo, C.; Hu, J. Effect of Thermal Extraction on Coal-Based Activated Carbon for Methane Decomposition to Hydrogen. ACS Omega 2020, 5, 2465–2472. [Google Scholar] [CrossRef]
- Wang, J.; Jin, L.; Li, Y.; Wang, M.; Hu, H. Effect of hydrogen additive on methane decomposition to hydrogen and carbon over activated carbon catalyst. Int. J. Hydrogen Energy 2018, 43, 17611–17619. [Google Scholar] [CrossRef]
- Gadkari, S.; Fidalgo, B.; Gu, S. Numerical analysis of microwave assisted thermocatalytic decomposition of methane. Int. J. Hydrogen Energy 2017, 42, 4061–4068. [Google Scholar] [CrossRef] [Green Version]
- Abanades, S.; Kimura, H.; Otsuka, H. Kinetic investigation of carbon-catalyzed methane decomposition in a thermogravimetric solar reactor. Int. J. Hydrogen Energy 2015, 40, 10744–10755. [Google Scholar] [CrossRef]
- Kameya, Y.; Hanamura, K. Variation in catalytic activity of carbon black during methane decomposition: Active site estimations from surface structural characteristics. Catal. Lett. 2012, 142, 460–463. [Google Scholar] [CrossRef]
- Lee, S.C.; Seo, H.J.; Han, G.Y. Hydrogen production by catalytic decomposition of methane over carbon black catalyst at high temperatures. Korean J. Chem. Eng. 2013, 30, 1716–1721. [Google Scholar] [CrossRef]
- Kameya, Y.; Hanamura, K. Kinetic and Raman spectroscopic study on catalytic characteristics of carbon blacks in methane decomposition. Chem. Eng. J. 2011, 173, 627–635. [Google Scholar] [CrossRef]
- Yoon, S.H.; Park, N.K.; Lee, T.J.; Yoon, K.J.; Han, G.Y. Hydrogen production by thermocatalytic decomposition of butane over a carbon black catalyst. Catal. Today 2009, 146, 202–208. [Google Scholar] [CrossRef]
- Yun, Y.H.; Lee, S.C.; Jang, J.T.; Yoon, K.J.; Bae, J.W.; Han, G.Y. Thermo-catalytic decomposition of propane over carbon black in a fluidized bed for hydrogen production. Int. J. Hydrogen Energy 2014, 39, 14800–14807. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, S.Y.; Kwak, J.H.; Han, G.Y.; Yoon, K.J. Hydrogen production by decomposition of ethane-containing methane over carbon black catalysts. Korean J. Chem. Eng. 2011, 28, 1833–1838. [Google Scholar] [CrossRef]
- Malaika, A.; Kozłowski, M. Hydrogen production by propylene-assisted decomposition of methane over activated carbon catalysts. Int. J. Hydrogen Energy 2010, 35, 10302–10310. [Google Scholar] [CrossRef]
- Hoffman, W.P.; Vastola, F.J.; Walker, P.L. Pyrolysis of propylene over carbon active sites—I: Kinetics. Carbon 1985, 23, 151–161. [Google Scholar] [CrossRef]
- Walker, P.L. Carbon: An old but new material revisited. Carbon 1990, 28, 261–279. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Chen, H.; Qi, M.; Zhang, G.; Hu, H.; Ma, X. Hydrogen production by catalytic methane decomposition: Carbon materials as catalysts or catalyst supports. Int. J. Hydrogen Energy 2017, 42, 19755–19775. [Google Scholar] [CrossRef]
- Yang, L.; Liu, F.; Liu, Y.; Quan, W.; He, J. Deep regeneration of activated carbon catalyst and autothermal analysis for chemical looping methane thermo-catalytic decomposition process. Int. J. Hydrogen Energy 2018, 43, 17633–17642. [Google Scholar] [CrossRef]
- Adamska, A.; Malaika, A.; Kozłowski, M. Carbon-catalyzed decomposition of methane in the presence of carbon dioxide. Energy Fuels 2010, 24, 3307–3312. [Google Scholar] [CrossRef]
- Abbas, H.F.; Daud, W.W. An experimental investigation into the CO2 gasification of deactivated activated-carbon catalyst used for methane decomposition to produce hydrogen. Int. J. Hydrogen Energy 2010, 35, 141–150. [Google Scholar] [CrossRef]
- Abbas, H.F.; Daud, W.W. Thermocatalytic decomposition of methane for hydrogen production using activated carbon catalyst: Regeneration and characterization studies. Int. J. Hydrogen Energy 2009, 34, 8034–8045. [Google Scholar] [CrossRef]
- Parkinson, B.; Balcombe, P.; Speirs, J.F.; Hawkes, A.D.; Hellgardt, K. Levelized cost of CO2 mitigation from hydrogen production routes. Energy Environ. Sci. 2019, 12, 19–40. [Google Scholar] [CrossRef]
- Weger, L.; Abánades, A.; Butler, T. Methane cracking as a bridge technology to the hydrogen economy. Int. J. Hydrogen Energy 2017, 42, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Keipi, T.; Tolvanen, K.E.; Tolvanen, H.; Konttinen, J. Thermo-catalytic decomposition of methane: The effect of reaction parameters on process design and the utilization possibilities of the produced carbon. Energy Convers. Manag. 2016, 126, 923–934. [Google Scholar] [CrossRef]
- Keipi, T.; Tolvanen, H.; Konttinen, J. Economic analysis of hydrogen production by methane thermal decomposition: Comparison to competing technologies. Energy Convers. Manag. 2018, 159, 264–273. [Google Scholar] [CrossRef]
- Abánades, A. Natural gas decarbonization as tool for Greenhouse Gases Emission Control. Front. Energy Res. 2018, 6, 47. [Google Scholar] [CrossRef]
- Srilatha, K.; Bhagawan, D.; Kumar, S.S.; Himabindu, V. Sustainable fuel production by thermocatalytic decomposition of methane–A review. S. Afr. J. Chem. Eng. 2017, 24, 156–167. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vander Wal, R.; Makiesse Nkiawete, M. Carbons as Catalysts in Thermo-Catalytic Hydrocarbon Decomposition: A Review. C 2020, 6, 23. https://doi.org/10.3390/c6020023
Vander Wal R, Makiesse Nkiawete M. Carbons as Catalysts in Thermo-Catalytic Hydrocarbon Decomposition: A Review. C. 2020; 6(2):23. https://doi.org/10.3390/c6020023
Chicago/Turabian StyleVander Wal, Randy, and Mpila Makiesse Nkiawete. 2020. "Carbons as Catalysts in Thermo-Catalytic Hydrocarbon Decomposition: A Review" C 6, no. 2: 23. https://doi.org/10.3390/c6020023