
Nanopore-Sequencing Metabarcoding for Identification of Phytopathogenic and Endophytic Fungi in Olive (Olea europaea) Twigs

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Growth, and Morphological Characterization of Fungal Isolates
2.3. DNA Extraction
2.4. Fungal-Specific Primers for Metabarcoding Assays
2.5. Verticillium dahliae-Specific Amplification in Plant Tissues by PCR
2.6. DNA Library Preparation and Sequencing
2.7. Data Analysis and Statistics
2.8. Data Availability
3. Results
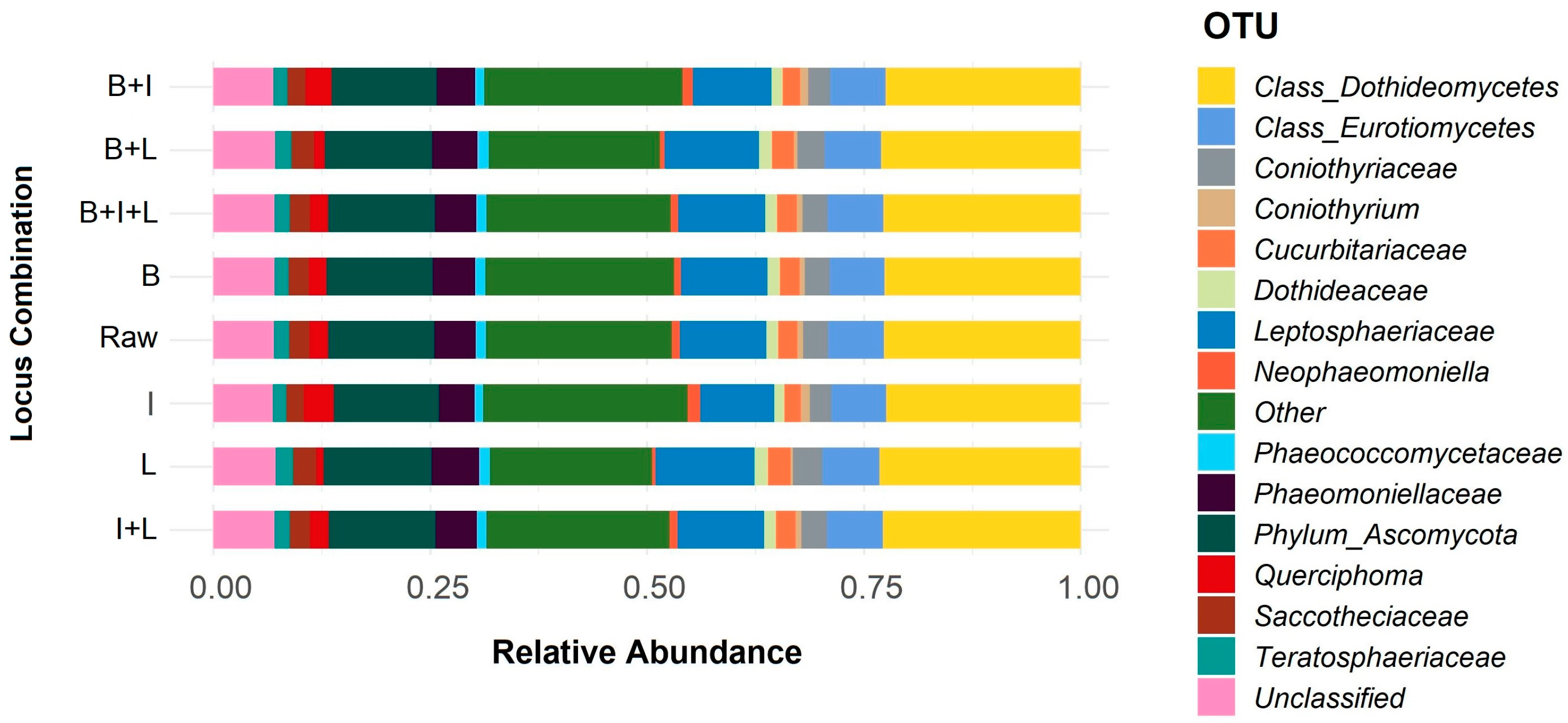
3.1. Selection and Verification of Plant Material and Fungi-Specific Primers for Metabarcoding
3.2. Metabarcoding Analysis of Healthy Olive Twigs
3.3. Metabarcoding Analysis of Symptomatic Olive Twigs
3.4. Data Analysis Using the BugSeq Platform
4. Discussion
4.1. Approach Definition, Selection of Plant Material and Verification of Fungal-Specific Primers
4.2. Initial Set Up of the ONT Fungi-Specific Multiplex Metabarcoding Method
4.3. Validation of the Metabarcoding Method on Infected Olive Twigs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aragona, M.; Haegi, A.; Valente, M.T.; Riccioni, L.; Orzali, L.; Vitale, S.; Luongo, L.; Infantino, A. New-Generation Sequencing Technology in Diagnosis of Fungal Plant Pathogens: A Dream Comes True? J. Fungi 2022, 8, 737. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Drenkhan, R.; Anslan, S.; Morales-Rodriguez, C.; Cleary, M. High-Throughput Identification and Diagnostics of Pathogens and Pests: Overview and Practical Recommendations. Mol. Ecol. Resour. 2019, 19, 47–76. [Google Scholar] [CrossRef]
- Antil, S.; Abraham, J.S.; Sripoorna, S.; Maurya, S.; Dagar, J.; Makhija, S.; Bhagat, P.; Gupta, R.; Sood, U.; Lal, R.; et al. DNA Barcoding, an Effective Tool for Species Identification: A Review. Mol. Biol. Rep. 2023, 50, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Gökdemir, F.Ş.; İşeri, Ö.D.; Sharma, A.; Achar, P.N.; Eyidoğan, F. Metagenomics Next Generation Sequencing (MNGS): An Exciting Tool for Early and Accurate Diagnostic of Fungal Pathogens in Plants. J. Fungi 2022, 8, 1195. [Google Scholar] [CrossRef]
- Liu, Y.X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A Practical Guide to Amplicon and Metagenomic Analysis of Microbiome Data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef]
- Coissac, E.; Riaz, T.; Puillandre, N. Bioinformatic Challenges for DNA Metabarcoding of Plants and Animals. Mol. Ecol. 2012, 21, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Navgire, G.S.; Goel, N.; Sawhney, G.; Sharma, M.; Kaushik, P.; Mohanta, Y.K.; Mohanta, T.K.; Al-Harrasi, A. Analysis and Interpretation of Metagenomics Data: An Approach. Biol. Proced. Online 2022, 24, 18. [Google Scholar] [CrossRef]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef]
- New, F.N.; Brito, I.L. What Is Metagenomics Teaching Us, and What Is Missed? Annu. Rev. Microbiol. 2020, 74, 117–135. [Google Scholar] [CrossRef]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.A.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J.; et al. Tree Lab: Portable Genomics for Early Detection of Plant Viruses and Pests in Sub-Saharan Africa. Genes 2019, 10, 632. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Kerkhof, L.J. Is Oxford Nanopore Sequencing Ready for Analyzing Complex Microbiomes? FEMS Microbiol. Ecol. 2021, 97, fiab001. [Google Scholar] [CrossRef] [PubMed]
- Leggett, R.M.; Clark, M.D. A World of Opportunities with Nanopore Sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef] [PubMed]
- Della Bartola, M.; Byrne, S.; Mullins, E. Characterization of Potato Virus y Isolates and Assessment of Nanopore Sequencing to Detect and Genotype Potato Viruses. Viruses 2020, 12, 478. [Google Scholar] [CrossRef]
- Chalupowicz, L.; Dombrovsky, A.; Gaba, V.; Luria, N.; Reuven, M.; Beerman, A.; Lachman, O.; Dror, O.; Nissan, G.; Manulis-Sasson, S. Diagnosis of Plant Diseases Using the Nanopore Sequencing Platform. Plant Pathol. 2019, 68, 229–238. [Google Scholar] [CrossRef]
- Fellers, J.P.; Webb, C.; Fellers, M.C.; Rupp, J.S.; De Wolf, E. Wheat Virus Identification within Infected Tissue Using Nanopore Sequencing Technology. Plant Dis. 2019, 103, 2199–2203. [Google Scholar] [CrossRef]
- Filloux, D.; Fernandez, E.; Loire, E.; Claude, L.; Galzi, S.; Candresse, T.; Winter, S.; Jeeva, M.L.; Makeshkumar, T.; Martin, D.P.; et al. Nanopore-Based Detection and Characterization of Yam Viruses. Sci. Rep. 2018, 8, 17879. [Google Scholar] [CrossRef]
- Liefting, L.W.; Waite, D.W.; Thompson, J.R. Application of Oxford Nanopore Technology to Plant Virus Detection. Viruses 2021, 13, 1424. [Google Scholar] [CrossRef]
- Sun, K.; Liu, Y.; Zhou, X.; Yin, C.; Zhang, P.; Yang, Q.; Mao, L.; Shentu, X.; Yu, X. Nanopore Sequencing Technology and Its Application in Plant Virus Diagnostics. Front. Microbiol. 2022, 13, 939666. [Google Scholar] [CrossRef]
- Waite, D.W.; Liefting, L.; Delmiglio, C.; Chernyavtseva, A.; Ha, H.J.; Thompson, J.R. Development and Validation of a Bioinformatic Workflow for the Rapid Detection of Viruses in Biosecurity. Viruses 2022, 14, 2163. [Google Scholar] [CrossRef]
- Faino, L.; Scala, V.; Albanese, A.; Modesti, V.; Grottoli, A.; Pucci, N.; Doddi, A.; L’Aurora, A.; Tatulli, G.; Reverberi, M.; et al. Nanopore Sequencing for the Detection and Identification of Xylella Fastidiosa Subspecies and Sequence Types from Naturally Infected Plant Material. Plant Pathol. 2021, 70, 1860–1870. [Google Scholar] [CrossRef]
- Llontop, M.E.M.; Sharma, P.; Flores, M.A.; Yang, S.; Pollok, J.; Tian, L.; Huang, C.; Rideout, S.; Heath, L.S.; Li, S.; et al. Strain-Level Identification of Bacterial Tomato Pathogens Directly from Metagenomic Sequences. Phytopathology 2020, 110, 768–779. [Google Scholar] [CrossRef]
- Hassan, A.H.; Bebawy, A.S.; Saad, M.T.; Mosaad, G.S.; Saad, B.T.; Eltayeb, W.N.; Aboshanab, K.M. Metagenomic Nanopore Sequencing versus Conventional Diagnosis for Identification of the Dieback Pathogens of Mango Trees. Biotechniques 2022, 73, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Badial, A.B.; Sherman, D.; Stone, A.; Gopakumar, A.; Wilson, V.; Schneider, W.; King, J. Nanopore Sequencing as a Surveillance Tool for Plant Pathogens in Plant and Insect Tissues. Plant Dis. 2018, 102, 1648–1652. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Costa, D.; Tavares, R.M.; Baptista, P.; Lino-Neto, T. Olive Fungal Epiphytic Communities Are Affected by Their Maturation Stage. Microorganisms 2022, 10, 376. [Google Scholar] [CrossRef]
- Costa, D.; Fernandes, T.; Martins, F.; Pereira, J.A.; Tavares, R.M.; Santos, P.M.; Baptista, P.; Lino-Neto, T. Illuminating Olea europaea L. Endophyte Fungal Community. Microbiol. Res. 2021, 245, 126693. [Google Scholar] [CrossRef]
- Hladnik, M.; Unković, N.; Janakiev, T.; Grbić, M.L.; Arbeiter, A.B.; Stanković, S.; Janaćković, P.; Gavrilović, M.; Rančić, D.; Bandelj, D.; et al. An Insight into an Olive Scab on the “Istrska Belica” Variety: Host-Pathogen Interactions and Phyllosphere Mycobiome. Microb. Ecol. 2022, 86, 1343–1363. [Google Scholar] [CrossRef]
- Brito, C.; Dinis, L.T.; Moutinho-Pereira, J.; Correia, C.M. Drought Stress Effects and Olive Tree Acclimation under a Changing Climate. Plants 2019, 8, 232. [Google Scholar] [CrossRef]
- Graniti, A.; Faedda, R.; Cacciola, S.O.; Magnano di San Lio, G. Olive Diseases in Changing Ecosystem. In Olive Diseases and Disorders; Transworld Research Network: Kerala, India, 2011. [Google Scholar]
- Ávila, A.; Groenewald, J.Z.; Trapero, A.; Crous, P.W. Characterisation and Epitypification of Pseudocercospora Cladosporioides, the Causal Organism of Cercospora Leaf Spot of Olives. Mycol. Res. 2005, 109, 881–888. [Google Scholar] [CrossRef]
- Moral, J.; Agustí-Brisach, C.; Raya, M.C.; Jurado-Bello, J.; López-Moral, A.; Roca, L.F.; Chattaoui, M.; Rhouma, A.; Nigro, F.; Sergeeva, V.; et al. Diversity of Colletotrichum Species Associated with Olive Anthracnose Worldwide. J. Fungi 2021, 7, 741. [Google Scholar] [CrossRef]
- Talhinhas, P.; Loureiro, A.; Oliveira, H. Olive Anthracnose: A Yield- and Oil Quality-Degrading Disease Caused by Several Species of Colletotrichum That Differ in Virulence, Host Preference and Geographical Distribution. Mol. Plant Pathol. 2018, 19, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Viruega, J.R.; Roca, L.F.; Moral, J.; Trapero, A. Factors Affecting Infection and Disease Development on Olive Leaves Inoculated with Fusicladium Oleagineum. Plant Dis. 2011, 95, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Buonaurio, R.; Almadi, L.; Famiani, F.; Moretti, C.; Agosteo, G.E.; Schena, L. Olive Leaf Spot Caused by Venturia Oleaginea: An Updated Review. Front. Plant Sci. 2023, 13, 1061136. [Google Scholar] [CrossRef]
- Sergeeva, V.; Nair, N.G.; Spooner-Hart, R. Evidence of Early Flower Infection in Olives (Olea europaea) by Colletotrichum Acutatum and C. Gloeosporioides Causing Anthracnose Disease. Australas. Plant Dis. Notes 2008, 3, 81–82. [Google Scholar] [CrossRef]
- Carroll, C.L.; Carter, C.A.; Goodhue, R.E.; Lawell, C.Y.C.L.; Subbarao, K.V. A Review of Control Options and Externalities for Verticillium Wilts. Phytopathology 2018, 108, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Montes-Osuna, N.; Mercado-Blanco, J. Verticillium Wilt of Olive and Its Control: What Did We Learn during the Last Decade? Plants 2020, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Ivic, D.; Ivanovic, A.; Milicevic, T.; Cvjetkovic, B. Shoot Necrosis of Olive Caused by Phoma Incompta, a New Disease of Olive in Croatia. Phytopathol. Mediterr. 2010, 49, 414–416. [Google Scholar]
- Rhouma, A.; Triki, M.A.; Krid, S.; Tuset, J.J.; Msallem, M. First Report of a Branch Dieback of Olive Trees in Tunisia Caused by a Phoma sp. Plant Dis. 2010, 94, 636. [Google Scholar] [CrossRef]
- Chliyeh, M.; Achbani, E.H.; Rhimini, Y.; Selmaoui, K.; Touhami, A.O.; Filali-Maltouf, A.; El Modafar, C.; Moukhli, A.; Oukabli, A.; Benkirane, R.; et al. Pathogenicity of Four Fungal Species on Fruits and Leaves of the Olive Tree (Olea europaea L.). Int. J. Pure Appl. Sci. 2014, 2, 1–9. [Google Scholar]
- Gharsallah, H.; Ksentini, I.; Frikha-Gargouri, O.; Hadj Taieb, K.; Ben Gharsa, H.; Schuster, C.; Chatti-kolsi, A.; Triki, M.A.; Ksantini, M.; Leclerque, A. Exploring Bacterial and Fungal Biodiversity in Eight Mediterranean Olive Orchards (Olea europaea L.) in Tunisia. Microorganisms 2023, 11, 1086. [Google Scholar] [CrossRef]
- Cobo-Díaz, J.F.; Baroncelli, R.; Le Floch, G.; Picot, A. Combined Metabarcoding and Co-Occurrence Network Analysis to Profile the Bacterial, Fungal and Fusarium Communities and Their Interactions in Maize Stalks. Front. Microbiol. 2019, 10, 261. [Google Scholar] [CrossRef] [PubMed]
- Walder, F.; Schlaeppi, K.; Wittwer, R.; Held, A.Y.; Vogelgsang, S.; Van Der Heijden, M.G.A. Community Profiling of Fusarium in Combination with Other Plant-Associated Fungi in Different Crop Species Using SMRT Sequencing. Front. Plant Sci. 2017, 8, 2019. [Google Scholar] [CrossRef]
- Behrens, F.H.; Fischer, M. Evaluation of Different Phyllosphere Sample Types for Parallel Metabarcoding of Fungi and Oomycetes in Vitis vinifera. Phytobiomes J. 2022, 6, 207–213. [Google Scholar] [CrossRef]
- Cureau, N.; Threlfall, R.; Savin, M.; Marasini, D.; Lavefve, L.; Carbonero, F. Year, Location, and Variety Impact on Grape-, Soil-, and Leaf-Associated Fungal Microbiota of Arkansas-Grown Table Grapes. Microb. Ecol. 2021, 82, 73–86. [Google Scholar] [CrossRef]
- Del Frari, G.; Gobbi, A.; Aggerbeck, M.R.; Oliveira, H.; Hansen, L.H.; Ferreira, R.B. Characterization of the Wood Mycobiome of Vitis Vinifera in a Vineyard Affected by Esca. Spatial Distribution of Fungal Communities and Their Putative Relation with Leaf Symptoms. Front. Plant Sci. 2019, 10, 910. [Google Scholar] [CrossRef] [PubMed]
- Morales-Cruz, A.; Figueroa-Balderas, R.; García, J.F.; Tran, E.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Profiling Grapevine Trunk Pathogens in Planta: A Case for Community-Targeted DNA Metabarcoding. BMC Microbiol. 2018, 18, 214. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Wisniewski, M.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Schena, L. Metagenomic Analysis of Fungal Diversity on Strawberry Plants and the Effect of Management Practices on the Fungal Community Structure of Aerial Organs. PLoS ONE 2016, 11, e0160470. [Google Scholar] [CrossRef]
- Kerdraon, L.; Barret, M.; Laval, V.; Suffert, F. Differential Dynamics of Microbial Community Networks Help Identify Microorganisms Interacting with Residue-Borne Pathogens: The Case of Zymoseptoria Tritici in Wheat. Microbiome 2019, 7, 125. [Google Scholar] [CrossRef]
- Ruiz Gómez, F.J.; Navarro-Cerrillo, R.M.; Pérez-de-Luque, A.; Oβwald, W.; Vannini, A.; Morales-Rodríguez, C. Assessment of Functional and Structural Changes of Soil Fungal and Oomycete Communities in Holm Oak Declined Dehesas through Metabarcoding Analysis. Sci. Rep. 2019, 9, 5315. [Google Scholar] [CrossRef]
- Shen, Z.; Penton, C.R.; Lv, N.; Xue, C.; Yuan, X.; Ruan, Y.; Li, R.; Shen, Q. Banana Fusarium Wilt Disease Incidence Is Influenced by Shifts of Soil Microbial Communities Under Different Monoculture Spans. Microb. Ecol. 2018, 75, 739–750. [Google Scholar] [CrossRef]
- Sugiyama, A.; Vivanco, J.M.; Jayanty, S.S.; Manter, D.K. Pyrosequencing Assessment of Soil Microbial Communities in Organic and Conventional Potato Farms. Plant Dis. 2010, 94, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, A.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Droby, S.; Schena, L. Metabarcoding Analysis of Fungal Diversity in the Phyllosphere and Carposphere of Olive (Olea europaea). PLoS ONE 2015, 10, e0131069. [Google Scholar] [CrossRef]
- Vergine, M.; Meyer, J.B.; Cardinale, M.; Sabella, E.; Hartmann, M.; Cherubini, P.; De Bellis, L.; Luvisi, A. The Xylella Fastidiosa-Resistant Olive Cultivar “Leccino” Has Stable Endophytic Microbiota during the Olive Quick Decline Syndrome (OQDS). Pathogens 2020, 9, 35. [Google Scholar] [CrossRef]
- Penland, M.; Mounier, J.; Pawtowski, A.; Tréguer, S.; Deutsch, S.M.; Coton, M. Use of Metabarcoding and Source Tracking to Identify Desirable or Spoilage Autochthonous Microorganism Sources during Black Olive Fermentations. Food Res. Int. 2021, 144, 110344. [Google Scholar] [CrossRef]
- Vita, F.; Sabbatini, L.; Sillo, F.; Ghignone, S.; Vergine, M.; Guidi Nissim, W.; Fortunato, S.; Salzano, A.M.; Scaloni, A.; Luvisi, A.; et al. Salt Stress in Olive Tree Shapes Resident Endophytic Microbiota. Front. Plant Sci. 2022, 13, 992395. [Google Scholar] [CrossRef] [PubMed]
- Malacrinò, A.; Mosca, S.; Li Destri Nicosia, M.G.; Agosteo, G.E.; Schena, L. Plant Genotype Shapes the Bacterial Microbiome of Fruits, Leaves, and Soil in Olive Plants. Plants 2022, 11, 613. [Google Scholar] [CrossRef]
- Mosca, S.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Schena, L. Molecular Analysis of Colletotrichum Species in the Carposphere and Phyllosphere of Olive. PLoS ONE 2014, 9, e114031. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Tooming-Klunderud, A.; Anslan, S. PacBio Metabarcoding of Fungi and Other Eukaryotes: Errors, Biases and Perspectives. New Phytol. 2018, 217, 1370–1385. [Google Scholar] [CrossRef]
- Raja, H.A.; Miller, A.N.; Pearce, C.J.; Oberlies, N.H. Fungal Identification Using Molecular Tools: A Primer for the Natural Products Research Community. J. Nat. Prod. 2017, 80, 756–770. [Google Scholar] [CrossRef]
- Carder, J.H.; Morton, A.; Tabrett, A.M.; Barbara, B.J. Detection and Differentiation by PCR of Subspecific Groups within Two Verticillium Species Causing Vascular Wilts in Herbaceous Hosts; Schots, A., Dewey, F.M., Oliver, R., Eds.; CAB International: Wallingford, UK, 1994. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Fan, J.; Huang, S.; Chorlton, S.D. BugSeq: A Highly Accurate Cloud Platform for Long-Read Metagenomic Analyses. BMC Bioinform. 2021, 22, 160. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Founda-Tion for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.r-project.org/ (accessed on 1 June 2023).
- Li, Y.; Steenwyk, J.L.; Chang, Y.; Wang, Y.; James, T.Y.; Stajich, J.E.; Spatafora, J.W.; Groenewald, M.; Dunn, C.W.; Hittinger, C.T.; et al. A Genome-Scale Phylogeny of the Kingdom Fungi. Curr. Biol. 2021, 31, 1653–1665.e5. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Leo Lahti, S.S.; Shetty, S.; Blake, T.; Salojarvi, J. Tools for Microbiome Analysis in R. Microbiome R Package. Available online: http://microbiome.github.io (accessed on 1 June 2023).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Francis, O.E.; Bendall, M.; Manimaran, S.; Hong, C.; Clement, N.L.; Castro-Nallar, E.; Snell, Q.; Schaalje, G.B.; Clement, M.J.; Crandall, K.A.; et al. Pathoscope: Species Identification and Strain Attribution with Unassembled Sequencing Data. Genome Res. 2013, 23, 1721–1729. [Google Scholar] [CrossRef]
- Martí, J.M. Recentrifuge: Robust Comparative Analysis and Contamination Removal for Metagenomics. PLoS Comput. Biol. 2019, 15, e1006967. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Yang, R.-H.; Su, J.-H.; Shang, J.-J.; Wu, Y.-Y.; Li, Y.; Bao, D.-P.I.; Yao, Y.-J. Evaluation of the Ribosomal DNA Internal Transcribed Spacer (ITS), Specifically ITS1 and ITS2, for the Analysis of Fungal Diversity by Deep Sequencing. PLOS ONE 2018, 13, e0206428. [Google Scholar] [CrossRef]
- Cui, C.; Herlihy, J.H.; Bombarely, A.; McDowell, J.M.; Haak, D.C. Draft Assembly of Phytophthora Capsici from Long-Read Sequencing Uncovers Complexity. Mol. Plant-Microbe Interact. 2019, 32, 1559–1563. [Google Scholar] [CrossRef]
- Krehenwinkel, H.; Pomerantz, A.; Henderson, J.B.; Kennedy, S.R.; Lim, J.Y.; Swamy, V.; Shoobridge, J.D.; Graham, N.; Patel, N.H.; Gillespie, R.G.; et al. Nanopore Sequencing of Long Ribosomal DNA Amplicons Enables Portable and Simple Biodiversity Assessments with High Phylogenetic Resolution across Broad Taxonomic Scale. Gigascience 2019, 8, giz006. [Google Scholar] [CrossRef]
- Phannareth, T.; Nunziata, S.O.; Stulberg, M.J.; Galvez, M.E.; Rivera, Y. Comparison of Nanopore Sequencing Protocols and Real-Time Analysis for Phytopathogen Diagnostics. Plant Health Prog. 2021, 22, 31–36. [Google Scholar] [CrossRef]
- Choudhary, P.; Singh, B.N.; Chakdar, H.; Saxena, A.K. DNA Barcoding of Phytopathogens for Disease Diagnostics and Bio-Surveillance. World J. Microbiol. Biotechnol. 2021, 37, 54. [Google Scholar] [CrossRef]
- Oliveira, M.; Azevedo, L. Molecular Markers: An Overview of Data Published for Fungi over the Last Ten Years. J. Fungi 2022, 8, 803. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Diaz, R.M.; Cirulli, M.; Bubici, G.; del Mar Jimenez-Gasco, M.; Antoniou, P.P.; Tjamos, E.C. Verticillium Wilt, a Major Threat to Olive Production: Current Status and Feature Prospects for Its Management. Plant Dis. 2012, 96, 304–329. [Google Scholar] [CrossRef] [PubMed]
- Da Lio, D.; Cobo-DÍaz, J.F.; Masson, C.; Chalopin, M.; Kebe, D.; Giraud, M.; Verhaeghe, A.; Nodet, P.; Sarrocco, S.; Le Floch, G.; et al. Combined Metabarcoding and Multi-Locus Approach for Genetic Characterization of Colletotrichum Species Associated with Common Walnut (Juglans Regia) Anthracnose in France. Sci. Rep. 2018, 8, 10765. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Liu, X.; Simeneh, Z.M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 Flow Cells in Single-Cell Whole-Genome Amplification and Whole-Genome Shotgun Sequencing. Comput. Struct. Biotechnol. J. 2023, 21, 2352–2364. [Google Scholar] [CrossRef]
- Kauserud, H. ITS Alchemy: On the Use of ITS as a DNA Marker in Fungal Ecology. Fungal Ecol. 2023, 65, 101274. [Google Scholar] [CrossRef]
- Morrison, G.A.; Fu, J.; Lee, G.C.; Wiederhold, N.P.; Cañete-Gibas, C.F.; Bunnik, E.M.; Wickes, B.L. Nanopore Sequencing of the Fungal Intergenic Spacer Sequence as a Potential Rapid Diagnostic Assay. J. Clin. Microbiol. 2020, 58, e01972-20. [Google Scholar] [CrossRef]
- Bautista-Jalón, L.S.; Frenkel, O.; Tsror, L.; Malcolm, G.M.; Gugino, B.K.; Lebiush, S.; Hazanovsky, M.; Milgroom, M.G.; Del Mar Jiménez-Gasco, M. Genetic Differentiation of Verticillium Dahliae Populations Recovered from Symptomatic and Asymptomatic Hosts. Phytopathology 2021, 111, 149–159. [Google Scholar] [CrossRef]
- Agustí-Brisach, C.; Jiménez-Urbano, J.P.; del Carmen Raya, M.; López-Moral, A.; Trapero, A. Vascular Fungi Associated with Branch Dieback of Olive in Super-High-Density Systems in Southern Spain. Plant Dis. 2021, 105, 797–818. [Google Scholar] [CrossRef]
- Nicoletti, R.; Di Vaio, C.; Cirillo, C. Endophytic Fungi of Olive Tree. Microorganisms 2020, 8, 1321. [Google Scholar] [CrossRef] [PubMed]
- Spies, C.F.J.; Mostert, L.; Carlucci, A.; Moyo, P.; van Jaarsveld, W.J.; du Plessis, I.L.; van Dyk, M.; Halleen, F. Dieback and Decline Pathogens of Olive Trees in South Africa. Persoonia Mol. Phylogeny Evol. Fungi 2020, 45, 196–220. [Google Scholar] [CrossRef] [PubMed]
- van Dyk, M.; Spies, C.F.J.; Mostert, L.; Halleen, F. Survey of Trunk Pathogens in South African Olive Nurseries. Plant Dis. 2021, 105, 1630–1639. [Google Scholar] [CrossRef]
- Vanga, B.R.; Panda, P.; Shah, A.S.; Thompson, S.; Woolley, R.H.; Ridgway, H.J.; Mundy, D.C.; Bulman, S. DNA Metabarcoding Reveals High Relative Abundance of Trunk Disease Fungi in Grapevines from Marlborough, New Zealand. BMC Microbiol. 2022, 22, 126. [Google Scholar] [CrossRef] [PubMed]
- Voglmayr, H.; Aguirre-Hudson, M.B.; Wagner, H.G.; Tello, S.; Jaklitsch, W.M. Lichens or Endophytes? The Enigmatic Genus Leptosillia in the Leptosilliaceae Fam. Nov. (Xylariales), and Furfurella Gen. Nov. (Delonicicolaceae). Persoonia Mol. Phylogeny Evol. Fungi 2019, 42, 228–260. [Google Scholar] [CrossRef] [PubMed]
- Bahri, H.; Ramos, V.; Mina, D.; Pereira, J.A.; Baptista, P. Characterization of Olive-Associated Fungi of Cultivars with Different Levels of Resistance to Anthracnose. Biol. Life Sci. Forum 2021, 4, 60. [Google Scholar]
- Chen, R.; Jiang, Y.M.; Wei, S.C.; Wang, Q.M. Kwoniella Shandongensis Sp. Nov., a Basidiomycetous Yeast Isolated from Soil and Bark from an Apple Orchard. Int. J. Syst. Evol. Microbiol. 2012, 62, 2774–2777. [Google Scholar] [CrossRef]
- Du, T.Y.; Karunarathna, S.C.; Zhang, X.; Dai, D.Q.; Mapook, A.; Suwannarach, N.; Xu, J.C.; Stephenson, S.L.; Elgorban, A.M.; Al-Rejaie, S.; et al. Endophytic Fungi Associated with Aquilaria Sinensis (Agarwood) from China Show Antagonism against Bacterial and Fungal Pathogens. J. Fungi 2022, 8, 1197. [Google Scholar] [CrossRef]
- Liu, X. Reinstatement of Coleonaema for Coleophoma Oleae and Notes on Coleophoma. Fungal Divers. 2016, 26, 187–204. [Google Scholar]
- Crous, P.W.; Schumacher, R.K.; Akulov, A.; Thangavel, R.; Hernández-Restrepo, M.; Carnegie, A.J.; Cheewangkoon, R.; Wingfield, M.J.; Summerell, B.A.; Quaedvlieg, W.; et al. New and Interesting Fungi. 2. Fungal Syst. Evol. 2019, 3, 57–134. [Google Scholar] [CrossRef] [PubMed]
- Crous, P.W.; Groenewald, J.Z. The Genera of Fungi—G 4: Camarosporium and Dothiora. IMA Fungus 2017, 8, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Li, M.; Huang, J.E.; Liu, F. Two New Species of Scolecobasidium (Venturiales, Sympoventuriaceae) Associated with True Mangrove Plants and S. Terrestre Comb. Nov. MycoKeys 2023, 96, 113–126. [Google Scholar] [CrossRef]
- Amiri, A.; Hawkins, A.W.; Mulvaney, K.A. Study of Fitness, Virulence, and Fungicide Sensitivity of Lambertella Corni-Maris Causing Yellow Rot on Apple. Plant Dis. 2017, 101, 738–743. [Google Scholar] [CrossRef]
- Jayawardena, R.S.; Hyde, K.D.; Chen, Y.J.; Papp, V.; Palla, B.; Papp, D.; Bhunjun, C.S.; Hurdeal, V.G.; Senwanna, C.; Manawasinghe, I.S.; et al. One Stop Shop IV: Taxonomic Update with Molecular Phylogeny for Important Phytopathogenic Genera: 76–100 (2020). Fungal Divers. 2020, 103, 87–218. [Google Scholar] [CrossRef]
- Crous, P.W.; Carnegie, A.J.; Wingfield, M.J.; Sharma, R.; Mughini, G.; Noordeloos, M.E.; Santini, A.; Shouche, Y.S.; Bezerra, J.D.P.; Dima, B.; et al. Fungal Planet Description Sheets: 868–950. Persoonia Mol. Phylogeny Evol. Fungi 2019, 42, 291–473. [Google Scholar] [CrossRef]
- Sakamoto, T.; Ortega, J.M. Taxallnomy: An Extension of NCBI Taxonomy That Produces a Hierarchically Complete Taxonomic Tree. BMC Bioinform. 2021, 22, 388. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A Comprehensive Update on Curation, Resources and Tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Sample | Region of Origin | Visual Symptoms | Plant Pathogenic Fungi Isolated | V. dahliae PCR Result | Selection for Metabarcoding |
|---|---|---|---|---|---|
| 1669 | Central Greece | yes | Verticillium dahliae | positive | yes |
| 1778 | Central Greece | yes | V. dahliae | positive | yes |
| 1939 | Peloponnese | yes | Alternaria sp., Cladosporium sp., Aspergillus sp., Penicillium sp. | negative | |
| 2136 | Western Greece | yes | Cladosporium sp., Cycloconium oleaginum, Penicillium sp. | negative | |
| 2179 | Attica | yes | Phoma sp., Cladosporium sp., Penicillium sp. | negative | yes |
| 2186 | Central Greece | yes | Cladosporium sp. | negative | |
| 2215 | Halkidiki | yes | Phoma sp., Cladosporium sp., C. oleaginum | negative | |
| 3100 | Attica | yes | V. dahliae | positive | yes |
| 3120 | Crete | no | none | negative | yes |
| 3121 | Crete | no | none | negative | |
| 3184 | Crete | yes | Phoma sp. | negative | yes |
| 3186 | Crete | no | none | negative | |
| 3869 | Peloponnese | yes | V. dahliae | positive | |
| 4500 | Western Greece | no | none | negative | |
| 4749 | Central Greece | no | none | negative |
| Plant Sample | DNA Sample | Primer Pairs Used in Individual PCRs | Barcode | Library | Sequencing Run |
|---|---|---|---|---|---|
| 3120 | 0 | ont-ITS1Fngs and ont-ITS4ngs | BR02 1 | L0 2 | 1 |
| ont-Bt2a and ont-Bt2b | |||||
| ont-ITS1Fngs and ont-LR5 | |||||
| 3120 | 1 3 | ont-ITS1Fngs and ont-ITS4ngs | BR07 | L1 | 2 |
| ont-Bt2a and ont-Bt2b | |||||
| 1669 | 2 | ont-ITS1Fngs and ont-ITS4ngs | BR08 | L2 | |
| ont-Bt2a and ont-Bt2b | |||||
| 1778 | 3 | ont-ITS1Fngs and ont-ITS4ngs | BR09 | L3 | |
| ont-Bt2a and ont-Bt2b | |||||
| 3100 | 4 | ont-ITS1Fngs and ont-ITS4ngs | BR10 | L4 | |
| ont-Bt2a and ont-Bt2b | |||||
| 2179 | 5 | ont-ITS1Fngs and ont-ITS4ngs | BR11 | L5 | |
| ont-Bt2a and ont-Bt2b | |||||
| 3184 | 6 | ont-ITS1Fngs and ont-ITS4ngs | BR12 | L6 | |
| ont-Bt2a and ont-Bt2b |
| OTU | ITS (I) | b-tub (B) | I + B | ol. 1 | OTU | ITS (I) | b-tub (B) | I + B | ol. |
|---|---|---|---|---|---|---|---|---|---|
| Alternaria | + 2 | + | (•) | Penicillium | + | + | + | (•) | |
| Aspergillus | + | (•) | Phaemoniella | + | (•) | ||||
| Aureobasidium | + | + | + | (•) | Phoma | + | (•) | ||
| Cladosporium | + | + | + | (•) | Podosphaera | + | + | ||
| Coleophoma | + | + | (•) | Pseudorobillarda | + | ||||
| Coniothyrium | + | + | (•) | Pseudocercospora | + | + | (•) | ||
| Constantinomyces | + | + | Pseudoseptoria | + | |||||
| Didymosphaeria | + | + | + | (•) | Pseudosydowia | + | |||
| Furfurella | + | + | + | (•) | Querciphoma | + | |||
| Fusarium | + | (•) | Scolecobasidium | + | |||||
| Hortaea | + | + | + | (•) | Septoria | + | (•) | ||
| Kwoniella | + | Stemphylium | + | (•) | |||||
| Lambertella | + | + | Stigmina | + | + | ||||
| Neocatenulostroma | + | (•) | Symmetrospora | + | + | (•) | |||
| Neocelosporium | + | Teratosphaeria | + | + | (•) | ||||
| Neodevriesia | + | (•) | Trimmatostroma | + | + | (•) | |||
| Neophaemoniella | + | + | (•) | Verticillium | + | + | + | (•) | |
| Ochrocladosporium | + | (•) | Xenocylindrosporium | + | (•) | ||||
| Paracamarosporium | + | C. ramotenellum | + | + | |||||
| Paraconiothyrium | + | + | (•) | P. brevicompactum | + | ||||
| Paracucurbitaria | + | + | (•) | V. dahliae | + | + | + | (•) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theologidis, I.; Karamitros, T.; Vichou, A.-E.; Kizis, D. Nanopore-Sequencing Metabarcoding for Identification of Phytopathogenic and Endophytic Fungi in Olive (Olea europaea) Twigs. J. Fungi 2023, 9, 1119. https://doi.org/10.3390/jof9111119
Theologidis I, Karamitros T, Vichou A-E, Kizis D. Nanopore-Sequencing Metabarcoding for Identification of Phytopathogenic and Endophytic Fungi in Olive (Olea europaea) Twigs. Journal of Fungi. 2023; 9(11):1119. https://doi.org/10.3390/jof9111119
Chicago/Turabian StyleTheologidis, Ioannis, Timokratis Karamitros, Aikaterini-Eleni Vichou, and Dimosthenis Kizis. 2023. "Nanopore-Sequencing Metabarcoding for Identification of Phytopathogenic and Endophytic Fungi in Olive (Olea europaea) Twigs" Journal of Fungi 9, no. 11: 1119. https://doi.org/10.3390/jof9111119