1. Introduction
Microorganisms possess an impressive variety of cellular mechanisms evolved to overcome various types of environmental stress, provided that the stressogenic factors do not occur in too great intensity, amount, or concentration. Yet, if the harmful conditions surpass the innate capacity to cope, the extreme stress exposure may cause accumulation of an overwhelming sum of cellular damage, leading to an excessive death and a dramatic decrease in population size. Consequently, the reconstitution of the population density in the wake of a massive death is a critical task. Given the prevalence and importance of microorganisms, the knowledge of the mechanisms and cellular factors underpinning population recovery is, thus, crucial for any comprehensive understanding of the strategies by which microorganisms persist even in strongly fluctuating environments.
It is principally within this conceptual and theoretical framework that our recent post-stress regrowth under starvation (RUS) studies have been developed [
1,
2,
3]. Namely, we use the model basidiomycotan fungus
Ustilago maydis to study the capacity of this single-celled haploid eukaryotic yeast-like microbe to reconstitute its populations after catastrophic stresses. To begin with, we employed the liquid holding (LH) assay system [
4,
5] to assess the ability of
U. maydis to recover from heavy oxidative insults, discovering that the fungus has a remarkable capacity to recover from massive damage [
1]. This initial investigation also established that the reconstitution of the devastated cell populations is promoted by growth and reproduction of the survivors, by feeding on the intracellular compounds leaked from the dead and dying cells. Importantly, the analysis of the growth effect of the substances released from the treated cells into the suspending medium revealed that the substrates have an opposing impact (nutritive and inhibitory) on the proliferation of freshly inoculated cells. On the one hand, the leakage products provide an accessible and rich supply of nutrients in quantities sufficient to support a robust multiplication of the inocula. However, increasing the dose of the stressors as well as prolonging the post-treatment incubation increases the inhibitory effect of the extracellular medium, which can be overcome by increasing the proportion of fresh to injured cells or by prolonging the time of LH incubation [
1]. The observations thus indicate that
U. maydis must possess and implement cellular operations involved not only in reabsorption of the released substrates but also in coping with their treatment-induced toxicity. Moreover, compared to
Saccharomyces cerevisiae,
U. maydis demonstrated evident superiority in effective processing and reuse of the leaked material, so that
S. cerevisiae actually resembled some of the
U. maydis RUS-mutants which were isolated as defective in performing RUS in peroxide-treated cell suspensions [
2].
There are several other interesting aspects of RUS and the reader is referred elsewhere for an account of a broader theoretical background to the formulation of the biological meaning as well as to the articulation of the challenges and questions related to this phenomenon [
3]. However, given that the exploration of RUS is still only in its pioneering phase, the elucidation of the molecular players and cellular operations involved in RUS is an issue that raises the most primary concerns. Indeed, it goes without saying that no microbial process can be adequately characterized, let alone understood, without actually furnishing sufficient information on the molecular underpinnings of the cellular operations supporting the phenomenon; at the very least, (micro)biological understanding requires knowing the factors involved. Therefore, the central focus of this study will be the identification of new cellular factors underlining RUS.
For precisely the same reasons, we have previously developed a screen for mutants defective in RUS, validated the screen by isolating a number of candidates, and characterized four of them (
adr1,
did4,
kel1,
tbp1) in considerable detail [
1]. However, since the method employed in screening the mutants did involve multiple experimental steps, such as treating suspensions of cells of each individual candidate with different doses of peroxide, followed by determination of the surviving fractions for each of the reactions both on immediate plating as well as periodically over 3 days of LH incubation, the isolation of these mutants was a time-consuming and laborious process. Therefore, to be more effective we changed to using the supernatants derived from heavily treated (1% H
2O
2) cell suspensions as the substrate for a phenotypic screen for
U. maydis mutants defective in RUS. This new, streamlined procedure was more efficient (less effort and time-consuming) than the previous one and resulted in the isolation of 33 mutants out of thousands readily screened. We have identified five of the mutants (
snf8,
slm1,
vrg4,
snf5,
hsf1) and partially characterized their phenotypes. In addition to being necessary for recycling of the damaged intracellular compounds, Slm1, Snf8, and Hsf1 are also important for protecting the
U. maydis genome against various genotoxins such as Ultravioletlight (UV), methyl methanesulfonate (MMS), and hydroxyurea (HU).
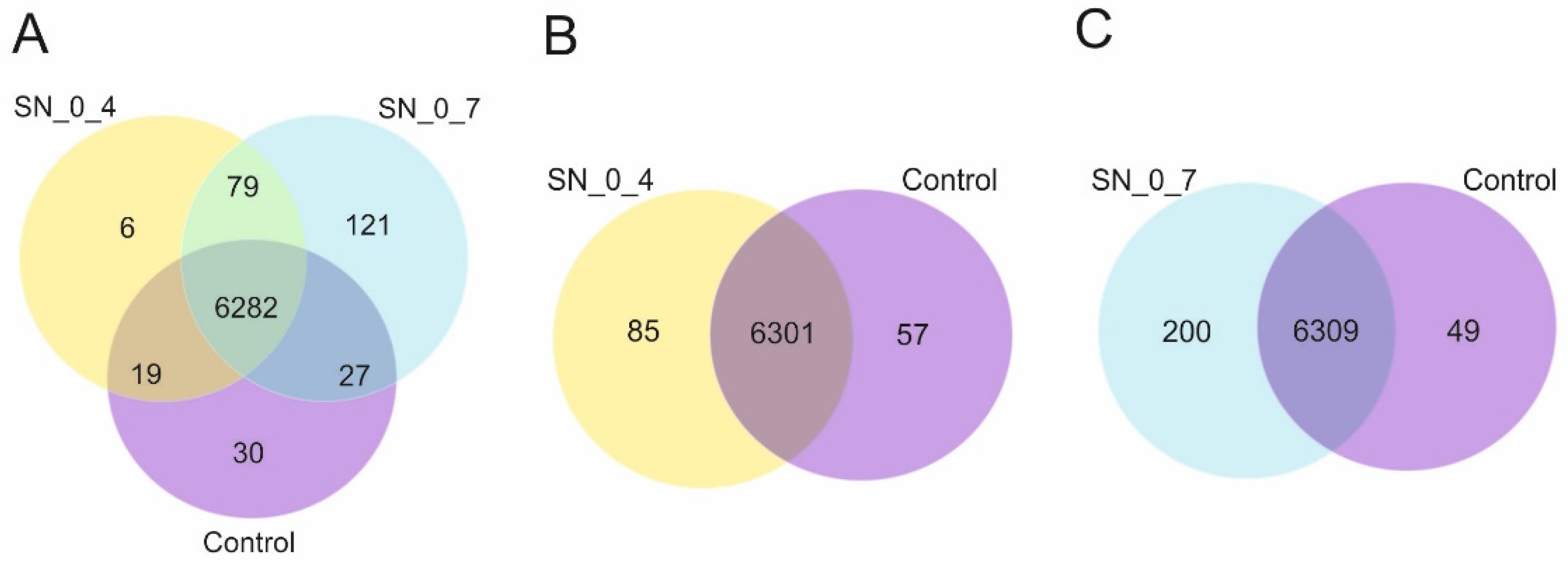
The investigation of the molecular factors involved in RUS may also be advanced and become more informative by the use of transcriptomics. So, we have performed global gene expression analysis through differential gene expression profiling of cells incubated in the supernatants derived from peroxide-treated cell suspensions vs. cells incubated in the rich growth medium. The transcriptome profiling revealed sets of uniquely expressed genes, and there was a positive correlation between the number of the exclusively expressed genes and the increasing toxicity of the substrates. Interestingly, among these genes was
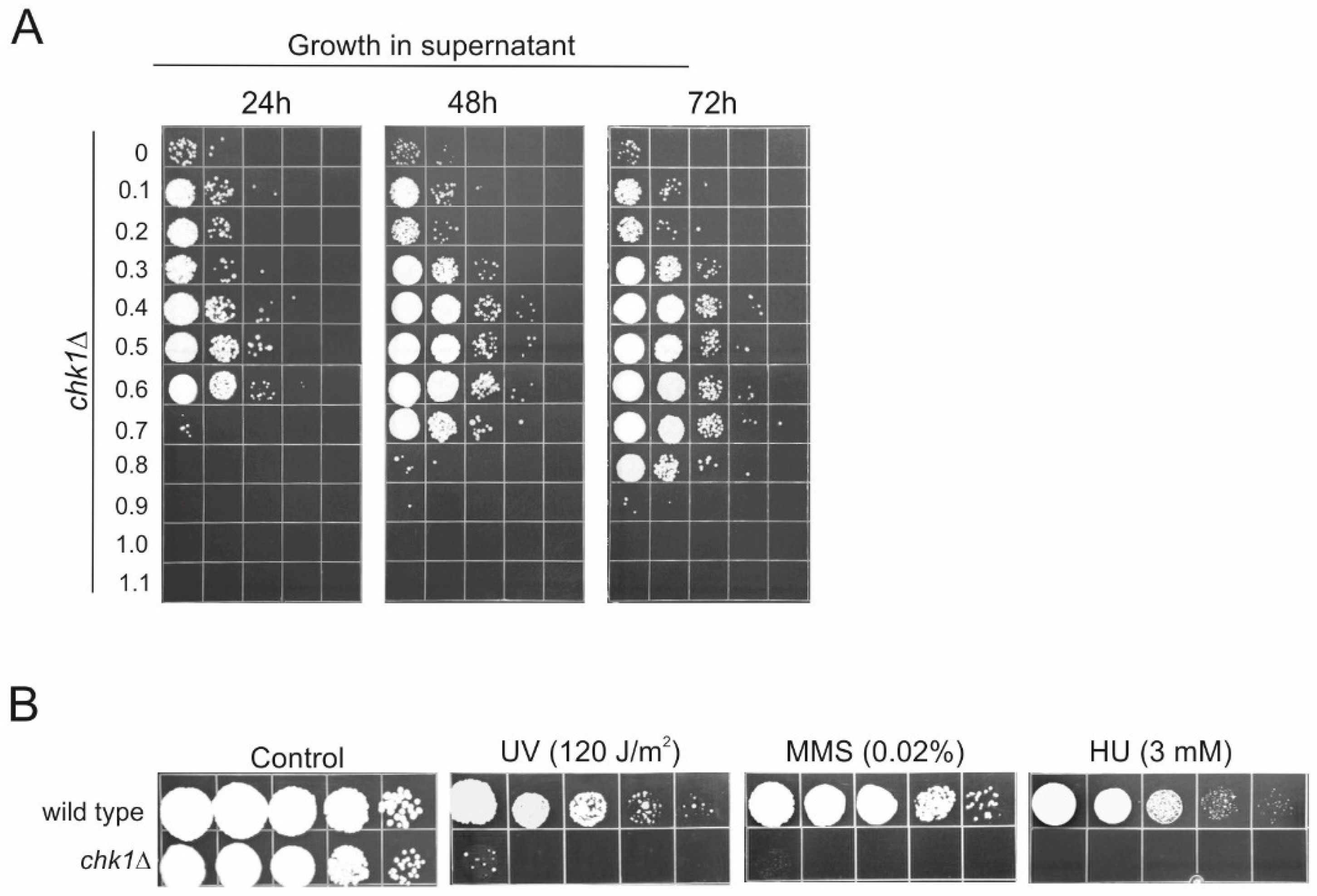
Chk1, encoding a protein kinase required for checkpoint-mediated cell cycle arrest, whose essential role in the maintenance of genome integrity has been extensively documented [
6,
7]. The ensuing gene function analysis of the
chk1 deletion mutant demonstrated very clearly that the gene is indispensable for the efficient growth of
U. maydis cells on the substrates freed from the cells killed by oxidation.
Overall, by applying these two approaches, we identified a set of cellular factors, of which some are of unknown functions or with predicted roles in different cellular processes such as cell cycle regulation, stress response, endomembrane trafficking, etc. Again, overlapping roles with maintenance of genome integrity have been observed for some of the identified factors. In any case, the current study provides a new foundation for future efforts to understand the functional roles of these factors in RUS.
2. Materials and Methods
2.1. U. maydis Growth and Peroxide Treatment
Culture methods, transformations, gene transfer, and treatments of
U. maydis have been described previously [
8,
9]. Nominal wild type strain UIMG10 (
nar1-6 a1b1) was used for mutagenesis, treatments with clastogenes, and genetic analysis [
10].
Cell number in liquid cultures was determined under microscope using a hemocytometer.
U. maydis was grown in rich YEPS medium (1% yeast extract, 2% sucrose, and 2% peptone) at 30 °C with agitation at 200 rpm. Growth rates of wild type and mutants were determined as described in [
1]. The results were analyzed by Student’s
t-test using SPSS statistical software.
p-values < 0.05 were considered significant.
Peroxide treatment was performed as described in [
1]. Briefly, liquid cultures were washed three times with distilled water. An amount of 2 × 10
7/mL of the washed cells was resuspended in 10 mM Fe
3+-sodium EDTA and incubated at 30 °C for 10 min. Peroxide treatment was performed by adding H
2O
2 to initiate Fenton reaction, which was stopped after 10 min by adding distilled water and pelleted cells by centrifugation at 4 °C. After additional two washings, cells were resuspended in water at 2 × 10
7/mL and plated immediately on a rich medium to measure survival or incubated at 30 °C with agitation and plated after 24–72 h to measure recovery. Spot assays were performed by making serial 10-fold dilutions from the initial cell suspension of 2 × 10
7/mL and then spotting 10 µL aliquots of each dilution in sequence on solid medium. Plates were incubated for 3 days at 30 °C.
For the preparation of the supernatants, following the peroxide treatment, the cell suspensions were washed twice, resuspended in water at 2 × 107/mL, and incubated in capped flasks mounted on an oscillating shaker at 30 °C for 4 h (or 16 h for the preparation of supernatants for the mutant screen). The cells were pelleted by centrifugation and the supernatants were passed through 0.45 µm-filters (Millipore, Burlington, MA, USA).
2.2. Mutant Screen and Gene Cloning
Exponentially growing cells of strain UIMG10 were spread on solid medium and irradiated with 254 nm UV light to a survival frequency of about 0.05%. Colonies arising from 3500 mutagenized survivors were tested for the ability to grow after incubation for 48 h in the cell-free supernatant derived from treated UIMG10 cells with 1% H2O2 in the presence of 10 mM Fe3+-sodium EDTA.
Five candidates with a loss of ability to grow were chosen for additional study (we will use the symbol “mir” to denote these mutants and it stands for mutants in RUS). The gene altered in mir3 was cloned by functional complementation of the MMS hypersensitivity after introducing a genomic DNA library prepared in a self-replicating vector with a hygromycin resistance marker as described previously [
10]. mir2, mir7, mir27, and mir29 were cloned by three consecutive rounds of incubation in supernatant derived from treatment with 1% peroxide and incubated for a 48 h period in water. After each round of incubation in supernatant, the survivors were grown to saturation in a medium containing 100 µg/mL hygromycin. A total of 2 × 10
7 cells was collected and incubated again through a subsequent round. After the third round, plasmids were extracted from survivors and the termini of cloned fragments were sequenced using primers from the vector. Usually, the genomic fragments are 5–15 kb long. Identity of the fragments was determined by matching with
U. maydis genome sequence in the annotated JGI MycoCosm database (
https://mycocosm.jgi.doe.gov; accessed on 4 February 2022). Candidate genes were subcloned to a single open reading frame (ORF) and retested for complementation. By sequencing of candidates ORFs, specific PCR amplicons were generated on genomic DNA of individual mutants and identities of mutated genes were determined: mir2—UMAG_11539 (Snf8), mir3—UMAG_03678 (Slm1), mir7—UMAG_01062 (Vrg4), mir27—UMAG_04381 (Snf5), mir29—UMAG_10368 (Hsf1).
2.3. RNA Extraction
For the analysis of the transcriptome, wild-type UIMG10 strain was grown over night and cells were washed two times. An amount of 2 × 107 cells/mL was inoculated in 13 mL of YEPS, or cell-free supernatants derived from treatments with 0.4% or 0.7% peroxide, and inocula were incubated for 30 min at 30 °C with agitation. After that, the cells were collected and total RNA of each sample was extracted using the GeneJET RNA Purification kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions with some modifications. Briefly, to increase the quantity of extracted RNA, the collected cells were resuspended in Yeast lyzing buffer (1 M sorbitol, 0.1 M EDTA, pH 7.4) containing 20 mM DTT and Lyzing enzyme from Trichoderma harzianum (Sigma, St. Louis, MO, USA). Suspensions were incubated for 30 min at 30 °C with agitation. Further steps were done according to the manufacturer’s instructions. DNase treatment was performed using DNA-free DNase Treatment and Removal Kit (Ambion, Austin, TX, USA).
2.4. Transcriptome Analysis
The total RNA purity, concentration, and quality were determined by an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). mRNA enrichment, cDNA library preparation, 150 bp pair-end RNA-sequencing took place on an Illumina NovaSeq 6000, and data analyses were carried out by Novogene Bioinformatics Technology Co., Ltd., Beijing, China.
2.5. Data Analysis
Raw data were preprocessed to remove adapters, poly-N > 10% sequences and low-quality reads (Qscore of over 50% bases of the read ≤ 5) using the fastp tool. Clean data (clean reads) were obtained by removing reads containing adapter and poly-N sequences and reads with low quality from raw data. Clean data served for calculating of Q20, Q30, and GC content. All the downstream analyses were based on clean data with a high quality. Paired-end clean reads were mapped to the reference genome using HISAT2 software.
Featurecounts were used to count the reads mapped for each gene and expressed in FPKM (short for the expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced), taking into account the effects of both gene length and reads count mapped to the gene [
11].
Prior to differential gene expression analysis, for each sequenced library, the read counts were adjusted by Trimmed Mean of M- values (TMM) through one scaling normalized factor. Differential expression analysis of two conditions was performed using the EdgeR (without biological replicates) R package. The p values were adjusted using the Benjamini and Hochberg approach for controlling the false discovery rate. Genes with corrected p value < 0.005 and |log2(Fold Change)| > 1 found by edgeR were assigned as differentially expressed.
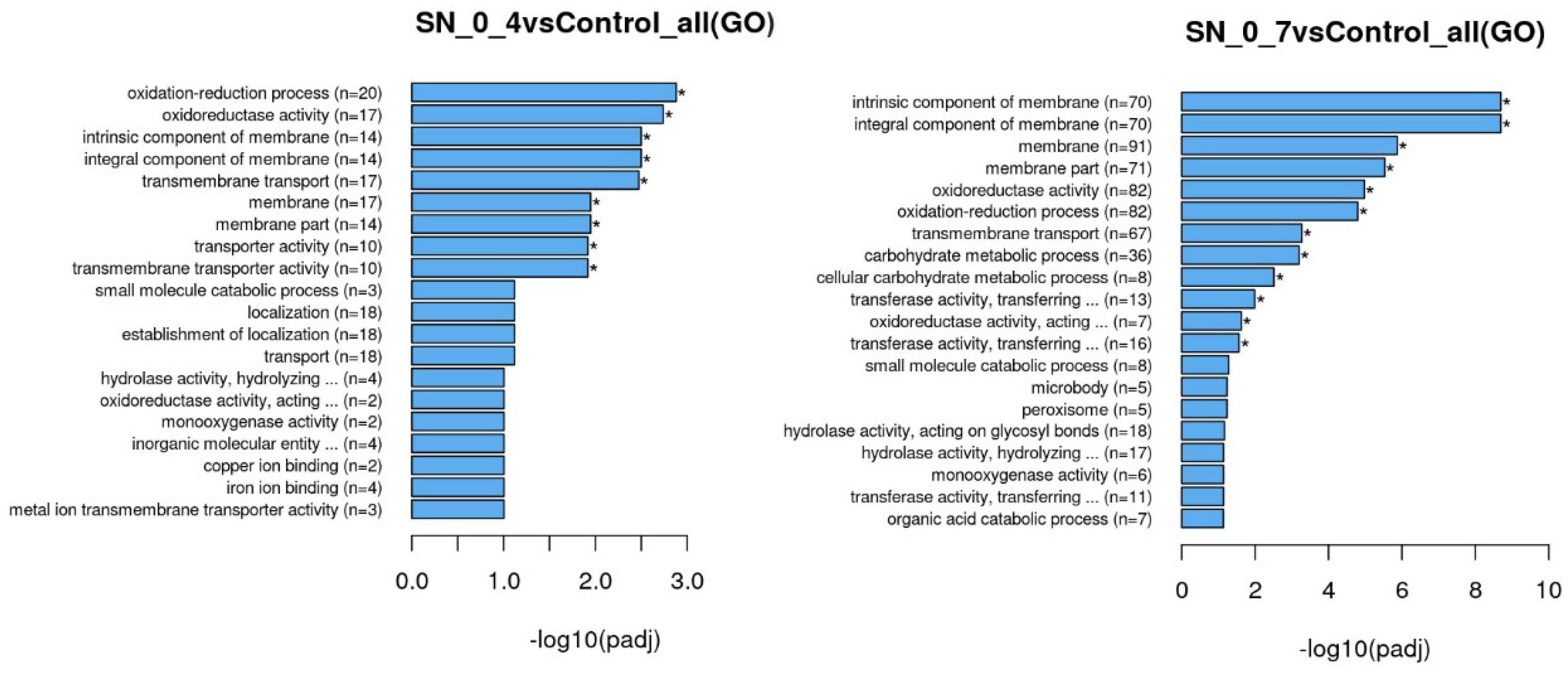
Enrichment analysis was performed using the clusterProfiler [
12]. R package (with correction of gene length bias and threshold of corrected
p value < 0.05) was used to test the statistical enrichment of differential expression genes in GO terms (Gene Ontology,
http://www.geneontology.org/; accessed on 9 May 2022) and KEGG pathways (Kyoto Encyclopedia of Genes and Genomes;
http://www.genome.jp/kegg/; accessed on 9 May 2022).
4. Discussion
This study was set in motion by the principal aspiration to improve biological understanding of RUS, which essentially requires further elucidation of the molecular basis of this phenomenon. Indeed, the identification of genes promoting the effective recycling of the damaged and liberated biomolecules is the key to any greater understanding of this major component of RUS. Therefore, to accomplish this objective we have undertaken a forward mutagenesis based on the new screen described above and carried out a comparative transcriptome analysis of U. maydis cells grown in YEPS and in the supernatants derived from peroxide-treated cell suspensions.
Employing the new screen, we isolated 33 mutants that display the RUS-deficient phenotype and so encode factors that may have roles in the processing and reusage of the toxic derivatives released from dying cells. We have identified five of the mutants (
snf8,
slm1,
vrg4,
snf5,
hsf1) and partially characterized their phenotypes. In addition to being required for recycling of the damaged and released intracellular compounds, Slm1, Snf8, and Hsf1 are also involved in the protection of the
U. maydis genome against various genotoxins such as UV, MMS, and HU. This is in line with our previous findings, which have found that two of the mutants isolated by the previous method (
did4 and
tbp1) exhibited a particularly strong DNA-repair phenotype [
1].
Prior to this investigation, we were operating with a rudimentary knowledge of the molecular players involved in RUS. Nevertheless, it was already apparent that the global cellular machinery required for RUS might actually be richly structured. Namely, the four cellular factors identified by our previous research (Adr1, Did4, Kel1, and Tbp1) had already been known to play roles in growth regulation, protein turnover, cytoskeleton structure, and transcription, indicating an assortment of the cellular functions underlying RUS. By now, these initial molecular insights are further exemplified by the variety of the molecular players identified by the new screen and, indeed, fully validated by the range of the cellular operations uncovered by the genome-wide transcriptome profiling (discussed below). Thus, RUS is evidently a very complex cellular operation that integrates a multiplicity of diverse molecular players and cellular processes. This conclusion is all the more in order given that our studies have identified several points of intersection between RUS and the maintenance of genome integrity. Of prime importance here is to elucidate the way in which these RUS factors contribute to the protection of the U. maydis genome.
The reciprocal angle of entry into this issue is from the perspective of genome protection. Namely, through the prism of transcriptomics and the subsequent gene-function analysis we have found that the checkpoint kinase 1 is indispensable for rendering
U. maydis cells effective in RUS. The finding that Chk1 is so importantly linked to RUS does indeed provide a reason for interrogating this connection further. Since Chk1 plays a major role in the checkpoint-mediated cell cycle arrest in response to DNA damage [
6,
7], the finding calls for a detailed investigation of the DNA repair factors (primarily those known to be under direct mediation by the Chk1 kinase) for their potential role in RUS. Given that the leakage from the dying cells may provide not only beneficial but also harmful (genotoxic) molecules—for instance, oxidized nucleotides—and given that the survivors must then cope with this challenge of energy-rich but risky (mutagenic?) compounds, it would certainly not be surprising if some of the DNA-repair proteins are required for the efficient RUS. To recap, whichever angle we viewed it from, the results suggest that RUS and the genome protection are linked into a unified operation that is an effective vehicle for regrowth/repopulation after devastating stress.
Of note, through transcriptome analysis, we found no changes of expression of any DNA-repair genes in cells grown in the SNs, except a gene for the putative 8-oxoguanine glycosylase involved in a base excision repair (UMAG_01304). Moreover, a decrease in the level of gene expression was detected for putative ATM (Ataxia Telangiectasia Mutated; Serine/Threonine Kinase) homolog (UMAG_15011) and putative Mlh1-DNA mismatch repair protein (UMAG_00274). Probably, the DNA-repair gene transcripts/proteins are present in cells at a sufficient level and change in transcriptome profiles is not the best way for detecting them, as has also been shown for
S. cerevisiae by [
22].
Returning to the theme of diversity of the cellular factors involved in RUS, the conserved factor Snf5 is a component of SWI/SNF chromatin remodeling complex important for chromatin structure and transcription regulation from a variety of promotors [
23,
24,
25]. With affected gene regulation, the yeast
snf5 null mutants display reduced growth on glucose and sucrose, are unable to grow on raffinose, galactose, or glycerol, and are hypersensitive to lithium and calcium ions [
26,
27,
28]. Additionally, Snf5 senses nucleocytoplasmic pH oscillations and induces transcriptional reprograming under carbon starvation [
29]. As a broad transcriptional regulator, it is not surprising that Snf5 is required for growth using damaged biomolecules. The same can be said of the hypothetical protein, homologous to Hsf1, a heat-shock transcription factor which is an activator of multiple genes in response to highly diverse stresses, including genes involved in protein folding, detoxification, energy generation, carbohydrate metabolism, and cell wall organization [
30,
31]. Importantly, the transcriptome analyses revealed seven heat shock proteins (Hsp 104, 16, 60, 70, 80, 90) upregulated in the higher-dosage SN, suggesting that the substrate-induced expression of these molecular chaperones and/or modifiers of proteostasis is at least partly responsible for enabling the cells for the optimal RUS. In other words, the upregulated expression of these factors that can elevate the cellular capacity for protein folding, preclude proteins from aggregating, facilitate aggregate dissolution, or suppress protein toxicity [
32,
33,
34] not only implies that the cells growing under RUS conditions do experience stress, but also suggests that they likely use the aggregate dissolution coupled with degradation to clear the cytoplasm of potentially toxic species. Further studies are required to address these possibilities. Interestingly, we found that transcription of the
Hsf1 gene, whose product in
S. cerevisiae activates transcription of a set of Hsp genes [
35,
36], was not affected during incubation in the suspensions. In both mutant hunts, through cell suspension and supernatants, we isolated mutants in endosomal trafficking, component of ESCRT-III (Did4), and component of ESCRT-II complex (Snf8/Vps22), respectively. Additionally, two genes coding for components of ESCRT-III (related to Snf7) were upregulated in cells grown in SN_0_7. This indicates that proper processing and recycling of retaken damaged compounds (for instance, recaptured oxidized peptides) is crucial for the multiplication on these harmful substrates. Similarly, endosome/multivesicular bodies formation and activities are necessary for the repair of membranes, which can also be critical for the stress-survivors to restore their membranes’ integrity after massive oxidative stress.
It is indeed worthy of note that actually none of the genes identified via previous or through the current screen for RUS-defective mutants were detected by our transcriptomic analysis as up- or downregulated. The simplest explanation for this would be that the genes are constitutively expressed. Another possible explanation is that the finding might be related to the time point chosen for the mRNA extraction. Namely, it is possible that some of the SN-responsive genes are induced only at later phases of growth in the SNs. Transcriptional analysis of
U. maydis genes at different time points during growth in the SNs will be useful in further understanding the global response in this RUS-competent organism. This is not a unique example of the lack of correlation between specific mutant fitness decrease and the increased gene expression. Comparison of expression and fitness profiling of yeast grown in altered environmental conditions (1 M NaCl, 1.5 M sorbitol, pH 8.0 and galactose) showed that less than 7% of the genes whose inactivation significantly affected growth in galactose-containing medium were also upregulated under the same conditions. Even more, in the case of pH 8.0, 1 M NaCl, and 1.5 M sorbitol, only 3.0%, 0.88%, and 0.34%, respectively, of the genes that showed a significant increase in mRNA expression also exhibited a significant decrease in fitness [
37,
38]. This can be the consequence of gene redundancy, and although many genes are upregulated, only a small fraction is essential for the process.
All this may suggest that the exclusive use of transcriptomic profiling to detect the genes underlying RUS may overlook the constitutively expressed regulatory genes that play crucial roles in establishing tolerance limits. Thus, like any other experimental approach, transcriptomics is associated with some limitations. On the other hand, we have a random mutagenesis and isolation of mutants methodology as one of the powerful tools for gene function discovery that provides several clear advantages. First of all, the functions of genes that are constitutively expressed can be evaluated. Another advantage is that mutational alternations tend to produce reliable phenotypic changes suitable for quantitative analysis. Additionally, a cause–effect relation between the mutation and the phenotype can even suggest mechanisms of the wild-type gene action. However, there are several disadvantages, too. First, the approach is less systematic and does fundamentally depend on chance. Another disadvantage is that essential genes may frequently be excluded. Additionally, given that the genes involved in RUS are scored by identifying loss of function mutations, any genes that have redundant functions are unlikely to be identified. Therefore, we would argue that the combination of these approaches would improve the outcomes. The results that have been accumulated in this study make it easier to agree.
Time-series gene expression profiling, expression of non-translated RNAs, changes in the proteome and metabolome profiles, post-translational modifications, and changes in protein localization could also be aspects of post-stress strategies of cell survival and can be the subject of some future investigations.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}