
Comparative Analysis of Transcriptomes of Ophiostoma novo-ulmi ssp. americana Colonizing Resistant or Sensitive Genotypes of American Elm

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Isolates
2.2. Inoculations and Recovery of Transcripts from Elm-Ophiostoma novo-ulmi Interaction
2.3. Ophiostoma novo-ulmi ssp. americana Transcriptome Mapping and Analysis
2.4. Functional Analysis of Candidate Pathogenicity Genes
3. Results
3.1. Overview of the O. novo-ulmi ssp. americana Transcriptome in Planta
3.2. Differentially Expressed Genes in O. novo-ulmi
3.3. Gene Expression Network Analysis
3.4. Functional Analysis of Candidate Pathogenicity Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwarz, M. Das zweigensterben der olmen, trauerweiden und pfirschbäume [The twig dying of the elms, willows, and peach trees]. Meded. Phytopath. Lab. Willie Commelin Sch. 1922, 5, 1–73. [Google Scholar]
- Brasier, C.M.; Kirk, S.A. Designation of the EAN and NAN Races of Ophiostoma novo-ulmi as Subspecies. Mycol. Res. 2001, 105, 547–554. [Google Scholar] [CrossRef]
- Brasier, C.M. Intercontinental Spread and Continuing Evolution of the Dutch Elm Disease Pathogens. In The Elms: Breeding, Conservation and Disease Management; Dunne, C.P., Ed.; Springer: Boston, MA, USA, 2000; pp. 61–72. [Google Scholar]
- Ganley, R.J.; Bulman, L.S. Dutch Elm Disease in New Zealand: Impacts from Eradication and Management Programmes. Plant Pathol. 2016, 65, 1047–1055. [Google Scholar] [CrossRef]
- Masuya, H.; Brasier, C.; Ichihara, Y.; Kubono, T.; Kanzaki, N. First Report of the Dutch Elm Disease Pathogens Ophiostoma ulmi and O. novo-ulmi in Japan. Plant Pathol. 2010, 59, 805. [Google Scholar] [CrossRef]
- Brasier, C.M.; Mehrotra, M.D. Ophiostoma himal-ulmi Sp. Nov., a New Species of Dutch Elm Disease Fungus Endemic to the Himalayas. Mycol. Res. 1995, 99, 205–215. [Google Scholar] [CrossRef]
- Khoshraftar, S.; Hung, S.; Khan, S.; Gong, Y.; Tyagi, V.; Parkinson, J.; Sain, M.; Moses, A.M.; Christendat, D. Sequencing and Annotation of the Ophiostoma ulmi Genome. BMC Genom. 2013, 14, 162. [Google Scholar] [CrossRef] [Green Version]
- Forgetta, V.; Leveque, G.; Dias, J.; Grove, D.; Lyons, R.; Genik, S.; Wright, C.; Singh, S.; Peterson, N.; Zianni, M.; et al. Sequencing of the Dutch Elm Disease Fungus Genome Using the Roche/454 GS-FLX Titanium System in a Comparison of Multiple Genomics Core Facilities. J. Biomol. Tech. 2013, 24, 39–49. [Google Scholar] [CrossRef]
- Bernier, L.; Aoun, M.; Bouvet, G.; Comeau, A.; Dufour, J.; Naruzawa, E.; Nigg, M.; Plourde, K. Genomics of the Dutch Elm Disease Pathosystem: Are We There Yet? IForest Biogeosci. For. 2015, 8, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Bernier, L. Genome-Wide Analysis of Parasitic Fitness Traits in a Non-Model Tree Pathogen. Can. J. Plant Pathol. 2016, 38, 153–163. [Google Scholar] [CrossRef]
- Comeau, A.M.; Dufour, J.; Bouvet, G.F.; Jacobi, V.; Nigg, M.; Henrissat, B.; Laroche, J.; Levesque, R.C.; Bernier, L. Functional Annotation of the Ophiostoma novo-ulmi Genome: Insights into the Phytopathogenicity of the Fungal Agent of Dutch Elm Disease. Genome Biol. Evol. 2015, 7, 410–430. [Google Scholar] [CrossRef] [Green Version]
- Hessenauer, P.; Fijarczyk, A.; Martin, H.; Prunier, J.; Charron, G.; Chapuis, J.; Bernier, L.; Tanguay, P.; Hamelin, R.C.; Landry, C.R. Hybridization and Introgression Drive Genome Evolution of Dutch Elm Disease Pathogens. Nat. Ecol. Evol. 2020, 4, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.; Faccoli, M. Dutch Elm Disease and Elm Bark Beetles: A Century of Association. IForest Biogeosci. For. 2015, 8, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Naruzawa, E.S.; Bernier, L. Control of Yeast-Mycelium Dimorphism In Vitro in Dutch Elm Disease Fungi by Manipulation of Specific External Stimuli. Fungal Biol. 2014, 118, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Hintz, W.; Pinchback, M.; de la Bastide, P.; Burgess, S.; Jacobi, V.; Hamelin, R.; Breuil, C.; Bernier, L. Functional Categorization of Unique Expressed Sequence Tags Obtained from the Yeast-like Growth Phase of the Elm Pathogen Ophiostoma novo-ulmi. BMC Genom. 2011, 12, 431. [Google Scholar] [CrossRef] [Green Version]
- Nigg, M.; Laroche, J.; Landry, C.R.; Bernier, L. RNAseq Analysis Highlights Specific Transcriptome Signatures of Yeast and Mycelial Growth Phases in the Dutch Elm Disease Fungus Ophiostoma novo-ulmi. G3 Genes Genomes Genet. 2015, 5, 2487–2495. [Google Scholar] [CrossRef] [Green Version]
- Nigg, M.; Bernier, L. From Yeast to Hypha: Defining Transcriptomic Signatures of the Morphological Switch in the Dimorphic Fungal Pathogen Ophiostoma novo-ulmi. BMC Genom. 2016, 17, 920. [Google Scholar] [CrossRef] [Green Version]
- Brasier, C. Some Genetical Aspects of Necrotrophy with Special Reference to Ophiostoma ulmi. In Genetics and Plant Pathogenesis; Day, P.R., Jellis, G.J., Eds.; Blackwell Scientific Publications: Oxford, UK, 1987; pp. 297–310. [Google Scholar]
- Takai, S.; Richards, W.C. Evidence for the Involvement of Cerato-Ulmin, the Ceratocystis ulmi Toxin, in the Development of Dutch Elm Disease. Physiol. Plant Pathol. 1983, 23, 275–280. [Google Scholar] [CrossRef]
- Temple, B.; Horgen, P.A.; Bernier, L.; Hintz, W.E. Cerato-Ulmin, a Hydrophobin Secreted by the Causal Agents of Dutch Elm Disease, Is a Parasitic Fitness Factor. Fungal Genet. Biol. 1997, 22, 39–53. [Google Scholar] [CrossRef]
- Bowden, C.; Smalley, E.; Guries, R.; Hubbes, M.; Temple, B.; Horgen, P. Lack of Association between Cerato-Ulmin Production and Virulence in Ophiostoma novo-ulmi. Mol. Plant. Microbe Interact. 1996, 9, 556–564. [Google Scholar] [CrossRef]
- Temple, B.; Bernier, L.; Hintz, W.E. Characterisation of the Polygalacturonase Gene of the Dutch Elm Disease Pathogen Ophiostoma novo-ulmi. N. Z. J. For. Sci. 2009, 39, 29–37. [Google Scholar]
- Et-Touil, A.; Brasier, C.M.; Bernier, L. Localization of a Pathogenicity Gene in Ophiostoma novo-ulmi and Evidence That It May Be Introgressed from O. ulmi. Mol. Plant-Microbe Interact. 1999, 12, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Plourde, K.; Bernier, L. An Ammonium Transporter Gene Is a Potential Pathogenicity Gene in Ophiostoma novo-ulmi (Abstract). Phytopathology 2006, 96, S93. [Google Scholar]
- Richards, W.C.; Takai, S.; Lin, D.; Hiratsuka, Y.; Asina, S. An Abnormal Strain of Ceratocystis ulmi Incapable of Producing External Symptoms of Dutch Elm Disease. Eur. J. For. Pathol. 1982, 12, 193–202. [Google Scholar] [CrossRef]
- Richards, W.C. Nonsporulation in the Dutch Elm Disease Fungus Ophiostoma ulmi: Evidence for Control by a Single Nuclear Gene. Can. J. Bot. 1994, 72, 461–467. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Carneiro, J.S.; de la Bastide, P.Y.; Chabot, M.; Lerch, L.; Hintz, W.E. Suppression of Polygalacturonase Gene Expression in the Phytopathogenic Fungus Ophiostoma novo-ulmi by RNA Interference. Fungal Genet. Biol. 2010, 47, 399–405. [Google Scholar] [CrossRef]
- Carneiro, J.S.; de la Bastide, P.Y.; Hintz, W.E. Regulated Gene Silencing in the Fungal Pathogen Ophiostoma novo-ulmi. Physiol. Mol. Plant Pathol. 2013, 82, 28–34. [Google Scholar] [CrossRef]
- Sarmiento-Villamil, J.L.; de Oliveira, T.C.; Naruzawa, E.S.; Bernier, L. An Efficient Strategy for Obtaining Mutants by Targeted Gene Deletion in Ophiostoma novo-ulmi. Front. Microbiol. 2021, 12, 699783. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Tanguay, P. CRISPR/Cas9 Gene Editing of the Dutch Elm Disease Pathogen Ophiostoma novo-ulmi. (Abstract). Can. J. Plant Pathol. 2019, 41, 163. [Google Scholar]
- Aoun, M.; Jacobi, V.; Boyle, B.; Bernier, L. Identification and Monitoring of Ulmus americana Transcripts during in Vitro Interactions with the Dutch Elm Disease Pathogen Ophiostoma novo-ulmi. Physiol. Mol. Plant Pathol. 2010, 74, 254–266. [Google Scholar] [CrossRef]
- Perdiguero, P.; Venturas, M.; Cervera, M.T.; Gil, L.; Collada, C. Massive Sequencing of Ulmus minor’s Transcriptome Provides New Molecular Tools for a Genus under the Constant Threat of Dutch Elm Disease. Front. Plant Sci. 2015, 6, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.T.; Coutin, J.F.; Shukla, M.; Dhaliwal, A.K.; Nigg, M.; Bernier, L.; Sherif, S.M.; Saxena, P.K. Deciphering the Genome-Wide Transcriptomic Changes during Interactions of Resistant and Susceptible Genotypes of American Elm with Ophiostoma novo-ulmi. J. Fungi 2022, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Sherif, S.M.; Shukla, M.R.; Murch, S.J.; Bernier, L.; Saxena, P.K. Simultaneous Induction of Jasmonic Acid and Disease-Responsive Genes Signifies Tolerance of American Elm to Dutch Elm Disease. Sci. Rep. 2016, 6, 21934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Dillies, M.A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, N.S.; Castel, D.; Estelle, J.; et al. A Comprehensive Evaluation of Normalization Methods for Illumina High-Throughput RNA Sequencing Data Analysis. Brief. Bioinform. 2013, 14, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. Wgcna: An r Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. Kegg: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Punt, P.J.; Dingemanse, M.A.; Kuyvenhoven, A.; Soede, R.D.M.; Pouwels, P.H.; van den Hondel, C.A.M.J.J. Functional Elements in the Promoter Region of the Aspergillus nidulans Gpda Gene Encoding Glyceraldehyde-3-Phosphate Dehydrogenase. Gene 1990, 93, 101–109. [Google Scholar] [CrossRef]
- Bernier, L.; Hubbes, M. Mutations in Ophiostoma ulmi Induced by N-Methyl-N′-Nitro-N-Nitrosoguanidine. Can. J. Bot. 1990, 68, 225–231. [Google Scholar] [CrossRef]
- Sarmiento-Villamil, J.L.; García-Pedrajas, N.E.; Cañizares, M.C.; García-Pedrajas, M.D. Molecular Mechanisms Controlling the Disease Cycle in the Vascular Pathogen Verticillium dahliae Characterized through Forward Genetics and Transcriptomics. Mol. Plant-Microbe Interact. 2020, 33, 825–841. [Google Scholar] [CrossRef]
- Paz, Z.; García-Pedrajas, M.D.; Andrews, D.L.; Klosterman, S.J.; Baeza-Montañez, L.; Gold, S.E. One Step Construction of Agrobacterium-Recombination-Ready-Plasmids (Oscar), an Efficient and Robust Tool for Atmt Based Gene Deletion Construction in Fungi. Fungal Genet. Biol. 2011, 48, 677–684. [Google Scholar] [CrossRef]
- Mullins, E.D.; Chen, X.; Romaine, P.; Raina, R.; Geiser, D.M.; Kang, S. Agrobacterium-Mediated Transformation of Fusarium oxysporum: An Efficient Tool for Insertional Mutagenesis and Gene Transfer. Phytopathology 2001, 91, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Brasier, C.M.; Webber, J.F. Positive Correlations between in Vitro Growth Rate and Pathogenesis in Ophiostoma ulmi. Plant Pathol. 1987, 36, 462–466. [Google Scholar] [CrossRef]
- Plourde, K.V.; Bernier, L. A Rapid Virulence Assay for the Dutch Elm Disease Fungus Ophiostoma novo-ulmi by Inoculation of Apple (Malus × Domestica ‘Golden Delicious’) Fruits. Plant Pathol. 2014, 63, 1078–1085. [Google Scholar] [CrossRef]
- Et-Touil, A.; Rioux, D.; Mathieu, F.M.; Bernier, L. External Symptoms and Histopathological Changes Following Inoculation of Elms Putatively Resistant to Dutch Elm Disease with Genetically Close Strains of Ophiostoma. Can. J. Bot. 2005, 83, 656–667. [Google Scholar] [CrossRef]
- Winnenburg, R. Phi-Base: A New Database for Pathogen Host Interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. Phi-Base: The Pathogen—Host Interactions Database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef] [PubMed]
- Sbaraini, N.; Andreis, F.C.; Thompson, C.E.; Guedes, R.L.M.; Junges, Â.; Campos, T.; Staats, C.C.; Vainstein, M.H.; Ribeiro de Vasconcelos, A.T.; Schrank, A. Genome-Wide Analysis of Secondary Metabolite Gene Clusters in Ophiostoma ulmi and Ophiostoma novo-ulmi Reveals a Fujikurin-like Gene Cluster with a Putative Role in Infection. Front. Microbiol. 2017, 8, 1063. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with Deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, G.H.; Savoldi, M.; Semighini, C.P.; De Oliveira, R.C.; Nunes, L.R.; Travassos, L.R.; Puccia, R.; Batista, W.L.; Ferreira, L.E.; Bogossian, A.P.; et al. Expressed Sequence Tag Analysis of the Human Pathogen Paracoccidioides brasiliensis Yeast Phase: Identification of Putative Homologues of Candida albicans Virulence and Pathogenicity Genes. Eukaryot. Cell 2003, 2, 34–48. [Google Scholar] [CrossRef] [Green Version]
- Thewes, S.; Kretschmar, M.; Park, H.; Schaller, M.; Filler, S.G.; Hube, B. In Vivo and Ex Vivo Comparative Transcriptional Profiling of Invasive and Non-Invasive Candida albicans Isolates Identifies Genes Associated with Tissue Invasion. Mol. Microbiol. 2007, 63, 1606–1628. [Google Scholar] [CrossRef]
- Felipe, M.S.S.; Torres, F.A.G.; Maranhao, A.Q.; Silva-Pereira, I.; Poças-Fonseca, M.J.; Campos, E.G.; Moraes, L.M.P.; Arraes, F.B.M.; Carvalho, M.J.A.; Andrade, R.V.; et al. Functional Genome of the Human Pathogenic Fungus Paracoccidioides brasiliensis. FEMS Immunol. Med. Microbiol. 2005, 45, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Felipe, M.S.S.; Andrade, R.V.; Arraes, F.B.M.; Nicola, A.M.; Maranhão, A.Q.; Torres, F.A.G.; Silva-Pereira, I.; Poças-Fonseca, M.J.; Campos, E.G.; Moraes, L.M.P.; et al. Transcriptional Profiles of the Human Pathogenic Fungus Paracoccidioides brasiliensis in Mycelium and Yeast Cells. J. Biol. Chem. 2005, 280, 24706–24714. [Google Scholar] [CrossRef] [Green Version]
- Cowen, L.E.; Lindquist, S. Hsp90 Potentiates the Rapid Evolution of New Traits: Drug Resistance in Diverse Fungi. Science 2005, 309, 2185–2189. [Google Scholar] [CrossRef]
- Cowen, L.E.; Singh, S.D.; Kohler, J.R.; Collins, C.; Zaas, A.K.; Schell, W.A.; Aziz, H.; Mylonakis, E.; Perfect, J.R.; Whitesell, L.; et al. Harnessing Hsp90 Function as a Powerful, Broadly Effective Therapeutic Strategy for Fungal Infectious Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 2818–2823. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.; Thakur, R.; Shankar, J.; Tiwari, S.; Thakur, R.; Shankar, J. Role of Heat-Shock Proteins in Cellular Function and in the Biology of Fungi. Biotechnol. Res. Int. 2015, 2015, 132635. [Google Scholar] [CrossRef] [Green Version]
- Jacobi, V.; Dufour, J.; Bouvet, G.F.; Aoun, M.; Bernier, L. Identification of Transcripts Up-Regulated in Asexual and Sexual Fruiting Bodies of the Dutch Elm Disease Pathogen Ophiostoma novo-ulmi. Can. J. Microbiol. 2010, 56, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Sützl, L.; Foley, G.; Gillam, E.M.J.; Bodén, M.; Haltrich, D. The Gmc Superfamily of Oxidoreductases Revisited: Analysis and Evolution of Fungal Gmc Oxidoreductases. Biotechnol. Biofuels 2019, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Segers, G.; Bradshaw, N.; Archer, D.; Blissett, K.; Oliver, R.P. Alcohol Oxidase Is a Novel Pathogenicity Factor for Cladosporium fulvum, but Aldehyde Dehydrogenase Is Dispensable. Mol. Plant-Microbe Interact. 2001, 14, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiGuistini, S.; Wang, Y.; Liao, N.Y.; Taylor, G.; Tanguay, P.; Feau, N.; Henrissat, B.; Chan, S.K.; Hesse-Orce, U.; Alamouti, S.M.; et al. Genome and Transcriptome Analyses of the Mountain Pine Beetle-Fungal Symbiont Grosmannia clavigera, a Lodgepole Pine Pathogen. Proc. Natl. Acad. Sci. USA 2011, 108, 2504–2509. [Google Scholar] [CrossRef] [Green Version]
- Douaiher, M.-N.; Nowak, E.; Durand, R.; Halama, P.; Reignault, P. Correlative Analysis of Mycosphaerella graminicola Pathogenicity and Cell Wall-Degrading Enzymes Produced in Vitro: The Importance of Xylanase and Polygalacturonase. Plant Pathol. 2007, 56, 79–86. [Google Scholar] [CrossRef]
- Kikot, G.E.; Hours, R.A.; Alconada, T.M. Contribution of Cell Wall Degrading Enzymes to Pathogenesis of Fusarium graminearum: A Review. J. Basic Microbiol. 2009, 49, 231–241. [Google Scholar] [CrossRef]
- Przybyl, K.; Dahm, H.; Ciesielska, A.; Molinski, K. Cellulolytic Activity and Virulence of Ophiostoma ulmi and O. novo-ulmi Isolates. For. Pathol. 2006, 36, 58–67. [Google Scholar] [CrossRef]
- Svaldi, R.; Elgersma, D.M. Further Studies on the Activity of Cell Wall Degrading Enzymes of Aggressive and Non-Aggressive Isolates of Ophiostoma ulmi. For. Pathol. 1982, 12, 29–36. [Google Scholar] [CrossRef]
- Binz, T.; Canevascini, G. Xylanases from the Dutch Elm Disease Pathogens Ophiostoma ulmi and Ophiostoma novo-ulmi. Physiol. Mol. Plant Pathol. 1996, 49, 159–175. [Google Scholar] [CrossRef]
- Scheffer, R.J.; Elgersma, D.M. A Scanning Electron Microscope Study of Cell Wall Degradation in Elm Wood by Aggressive and Non-Aggressive Isolates of Ophiostoma ulmi. For. Pathol. 1982, 12, 25–28. [Google Scholar] [CrossRef]
- Binz, T.; Canevascini, G. Purification and Partial Characterization of the Extracellular Laccase from Ophiostoma novo-ulmi. Curr. Microbiol. 1997, 35, 278–281. [Google Scholar] [CrossRef]
- Binz, T.; Canevascini, G. Differential Production of Extracellular Laccase in the Dutch Elm Disease Pathogens Ophiostoma ulmi and O. novo-ulmi. Mycol. Res. 1996, 100, 1060–1064. [Google Scholar] [CrossRef]
- Comménil, P.; Belingheri, L.; Bauw, G.; Dehorter, B. Molecular Characterization of a Lipase Induced in Botrytis cinerea by Components of Grape Berry Cuticle. Physiol. Mol. Plant Pathol. 1999, 55, 37–43. [Google Scholar] [CrossRef]
- Berto, P.; Comménil, P.; Belingheri, L.; Dehorter, B. Occurrence of a Lipase in Spores of Alternaria brassicicola with a Crucial Role in the Infection of Cauliflower Leaves. FEMS Microbiol. Lett. 1999, 180, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wang, F.; Liu, G.; Greenshields, D.; Shen, W.; Kaminskyj, S.; Hughes, G.R.; Peng, Y.; Selvaraj, G.; Zou, J.; et al. Analysis of a Blumeria graminis-Secreted Lipase Reveals the Importance of Host Epicuticular Wax Components for Fungal Adhesion and Development. Mol. Plant-Microbe Interact. 2009, 22, 1601–1610. [Google Scholar] [CrossRef] [Green Version]
- Claydon, N.; Elgersma, D.M.; Grove, J.F. The Phytotoxicity of Some Phenolic Metabolic Products of Ophiostoma ulmi to Ulmus sp. Neth. J. Plant Pathol. 1980, 86, 229–237. [Google Scholar] [CrossRef]
- Scheffer, R.J.; Liem, J.I.; Elgersma, D.M. Production in Vitro of Phytotoxic Compounds by Non-Aggressive and Aggressive Isolates of Ophiostoma ulmi, the Dutch Elm Disease Pathogen. Physiol. Mol. Plant Pathol. 1987, 30, 321–335. [Google Scholar] [CrossRef]
- Bhatnagar, D.; McCormick, S.P.; Lee, L.S.; Hill, R.A. Identification of O-Methylsterigmatocystin as an Aflatoxin B1 and G1 Precursor in Aspergillus parasiticus. Appl. Environ. Microbiol. 1987, 53, 1028–1033. [Google Scholar] [CrossRef] [Green Version]
- Detroy, R.W.; Lillehoj, E.B.; Ciegler, A. Aflatoxin and Related Compounds. In Microbial Toxins; Ciegler, A., Kadis, S., Ajl, S.J., Eds.; Academic Press: New York, NY, USA, 1971; pp. 3–178. [Google Scholar]
- Desjardins, A.E.; Hohn, T.M. Mycotoxins in Plant Pathogenesis. Mol. Plant-Microbe Interact. 1997, 10, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Chang, P.; Ehrlich, K.C.; Cary, J.W.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A.; Linz, J.E.; Woloshuk, C.P.; Bennett, J.W. Clustered Pathway Genes in Aflatoxin Biosynthesis. Appl. Environ. Microbiol. 2004, 70, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.M.; Howlett, B.J. Secondary Metabolism: Regulation and Role in Fungal Biology. Curr. Opin. Microbiol. 2008, 11, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Treutter, D. Significance of Flavonoids in Plant Resistance: A Review. Environ. Chem. Lett. 2006, 4, 147–157. [Google Scholar] [CrossRef]
- Ebel, J. Phytoalexin Synthesis: The Biochemincal Analysis of the Induction Process. Annu. Rev. Phytopathol. 1986, 24, 235–264. [Google Scholar] [CrossRef]
- Daayf, F.; El Hadrami, A.; El-Bebany, A.F.; Henriquez, M.A.; Yao, Z.; Derksen, H.; El-Hadrami, I.; Adam, L.R. Phenolic Compounds in Plant Defense and Pathogen Counter-Defense Mechanisms. In Recent Advances in Polyphenol Research; Cheynier, V., Sarni-Manchado, P., Quideau, S., Eds.; Wiley-Blackwell: Chichester, UK, 2012; Volume 3, pp. 191–208. [Google Scholar]
- Duchesne, L.C.; Jeng, R.S.; Hubbes, M. Accumulation of Phytoalexins in Ulmus americana in Response to Infection by a Nonaggressive and an Aggressive Strain of Ophiostoma ulmi. Can. J. Bot. 1985, 63, 678–680. [Google Scholar] [CrossRef]
- Dumas, M.T.; Strunz, G.M.; Hubbes, M.; Jeng, R.S. Isolation and Identification of Six Mansonones from Ulmus americana Infected with Ceratocystis ulmi. Experientia 1983, 39, 1089–1090. [Google Scholar] [CrossRef]
- Yang, D.; Hubbes, M.; Jeng, R.S. A Glycoprotein Isolated from Culture Filtrates of Ophiostoma ulmi as a Mansonone-Inducing Elicitor on Elm Callus. Mycol. Res. 1994, 98, 295–300. [Google Scholar] [CrossRef]
- Blomquist, G.J.; Figueroa-Teran, R.; Aw, M.; Song, M.; Gorzalski, A.; Abbott, N.L.; Chang, E.; Tittiger, C. Pheromone Production in Bark Beetles. Insect Biochem. Mol. Biol. 2010, 40, 699–712. [Google Scholar] [CrossRef]
- Rottava, I.; Cortina, P.F.; Zanella, C.A.; Cansian, R.L.; Toniazzo, G.; Treichel, H.; Antunes, O.A.C.; Oestreicher, E.G.; De Oliveira, D. Microbial Oxidation of (−)-a-Pinene to Verbenol Production by Newly Isolated Strains. Appl. Biochem. Biotechnol. 2010, 162, 2221–2231. [Google Scholar] [CrossRef]
- McLeod, G.; Gries, R.; von Reuss, S.H.; Rahe, J.E.; McIntosh, R.; König, W.A.; Gries, G. The Pathogen Causing Dutch Elm Disease Makes Host Trees Attract Insect Vectors. Proc. Biol. Sci. 2005, 272, 2499–2503. [Google Scholar] [CrossRef]
- Dai, L.; Li, Z.; Yu, J.; Ma, M.; Zhang, R.; Chen, H.; Pham, T. The CYP51F1 Gene of Leptographium qinlingensis: Sequence Characteristic, Phylogeny and Transcript Levels. Int. J. Mol. Sci. 2015, 16, 12014–12034. [Google Scholar] [CrossRef] [Green Version]
- Stringer, M.A.; Timberlake, W.E. Cerato-Ulmin, a Toxin Involved in Dutch Elm Disease, Is a Fungal Hydrophobin. Plant Cell 1993, 5, 145–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Sorbo, G.; Scala, F.; Parrella, G.; Lorito, M.; Comparini, C.; Ruocco, M.; Scala, A. Functional Expression of the Gene Cu, Encoding the Phytotoxic Hydrophobin Cerato-Ulmin, Enables Ophiostoma quercus, a Nonpathogen on Elm, to Cause Symptoms of Dutch Elm Disease. Mol. Plant-Microbe Interact. 2000, 13, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.G.; Garcia-Pedrajas, M.D.; Gold, S.E.; Perlin, M.H. Isolation and Characterization from Pathogenic Fungi of Genes Encoding Ammonium Permeases and Their Roles in Dimorphism. Mol. Microbiol. 2003, 50, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Marini, A.M.; Soussi-Boudekou, S.; Vissers, S.; Andre, B. A Family of Ammonium Transporters in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997, 17, 4282–4293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, K.; Morschhäuser, J. The Mep2p Ammonium Permease Controls Nitrogen Starvation-Induced Filamentous Growth in Candida albicans. Mol. Microbiol. 2005, 56, 649–669. [Google Scholar] [CrossRef]
- Et-Touil, A.; Dusabenyagasani, M.; Bouvet, G.F.; Brasier, C.M.; Bernier, L. Ophiostoma ulmi DNA Naturally Introgressed into an Isolate of Ophiostoma novo-ulmi Is Clustered around Pathogenicity and Mating Type Loci. Phytoprotection 2019, 99, 1–11. [Google Scholar] [CrossRef]
- Wang, C.; St Leger, R.J. The Mad1 Adhesin of Metarhizium anisopliae Links Adhesion with Blastospore Production and Virulence to Insects, and the Mad2 Adhesin Enables Attachment to Plants. Eukaryot. Cell 2007, 6, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Soundararajan, S.; Jedd, G.; Li, X.; Ramos-Pamploña, M.; Chua, N.H.; Naqvi, N.I. Woronin Body Function in Magnaporthe grisea Is Essential for Efficient Pathogenesis and for Survival during Nitrogen Starvation Stress. Plant Cell 2004, 16, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Gao, H.; Li, J.; Liu, L.; Liu, Z.; Zhang, K.-Q. The Woronin Body in the Nematophagous Fungus Arthrobotrys oligospora Is Essential for Trap Formation and Efficient Pathogenesis. Fungal Biol. 2017, 121, 11–20. [Google Scholar] [CrossRef]
- Royer, J.C.; Dewar, K.; Hubbes, M.; Horgen, P.A. Analysis of a High Frequency Transformation System for Ophiostoma ulmi, the Causal Agent of Dutch Elm Disease. Mol. Gen. Genet. 1991, 225, 168–176. [Google Scholar] [CrossRef]
- Denisov, Y.; Yarden, O.; Freeman, S. Impaired Purine Biosynthesis Affects Pathogenicity of Fusarium oxysporum f. sp. melonis. Eur. J. Plant Pathol. 2005, 112, 293–297. [Google Scholar] [CrossRef]
- Martín, J.A.; Domínguez, J.; Solla, A.; Brasier, C.M.; Webber, J.F.; Santini, A.; Martínez-Arias, C.; Bernier, L.; Gil, L. Complexities Underlying the Breeding and Deployment of Dutch Elm Disease Resistant Elms. New For. 2021, 1–36. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Valley Forge | Susceptible | |||
|---|---|---|---|---|
| 0 h | 96 h | 0 h | 96 h | |
| Total reads 1 | 75,742,361 | 73,144,879 | 66,441,814 | 75,518,322 |
| Reads fully mapped to O. novo-ulmi genes (exons + introns) | 0 | 24,193 | 0 | 65,910 |
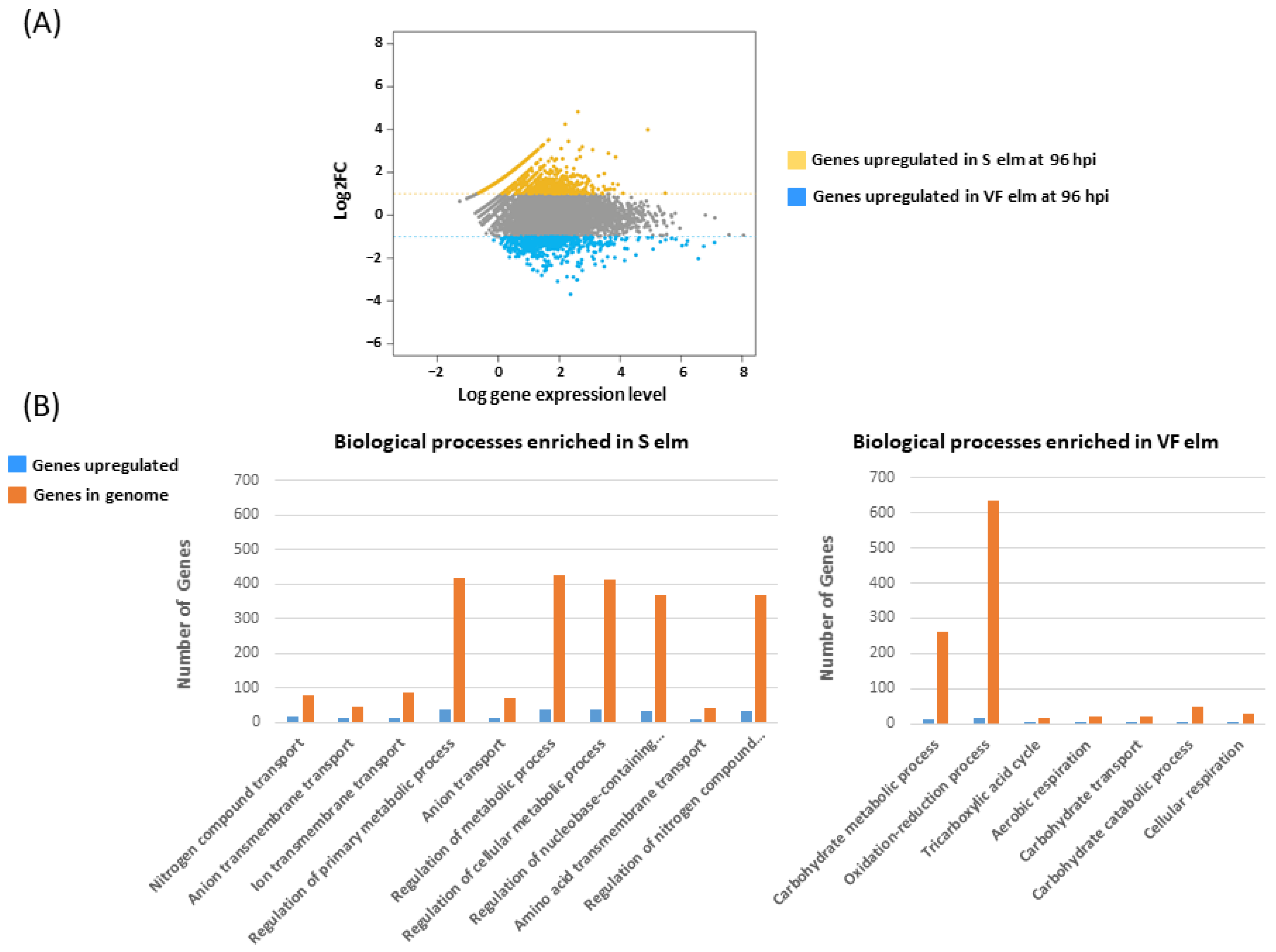
| Treatment | Expressed | Upregulated (logFC(1) 1) | Annotated Upregulated | CAZYmes Upregulated | Signal Peptides Upregulated | PHI-Base Upregulated |
|---|---|---|---|---|---|---|
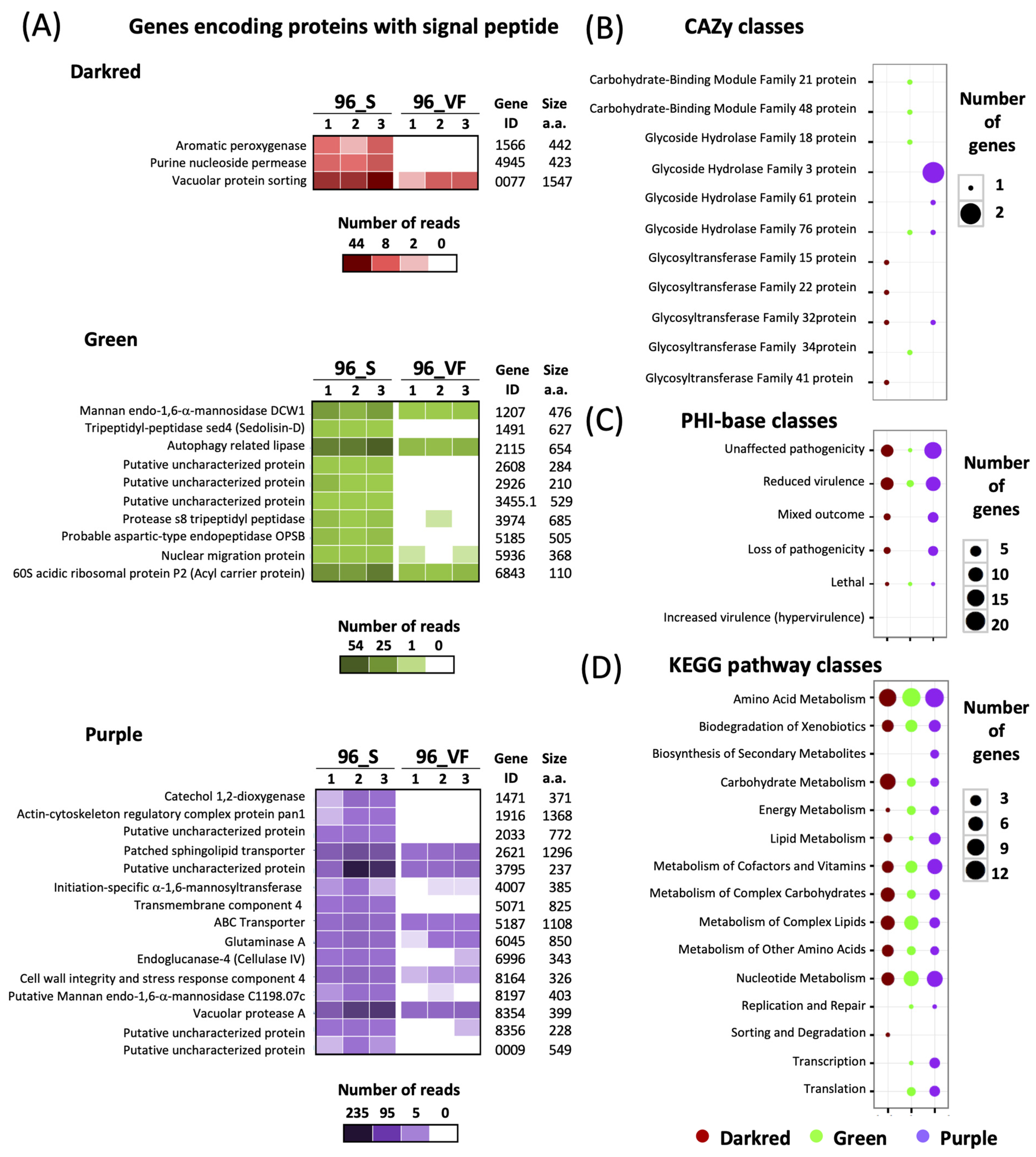
| 96h_S | 5392 | 370 | 303 | 16 | 20 | 94 |
| 96h_VF | 3014 | 102 | 84 | 17 | 26 | 29 |
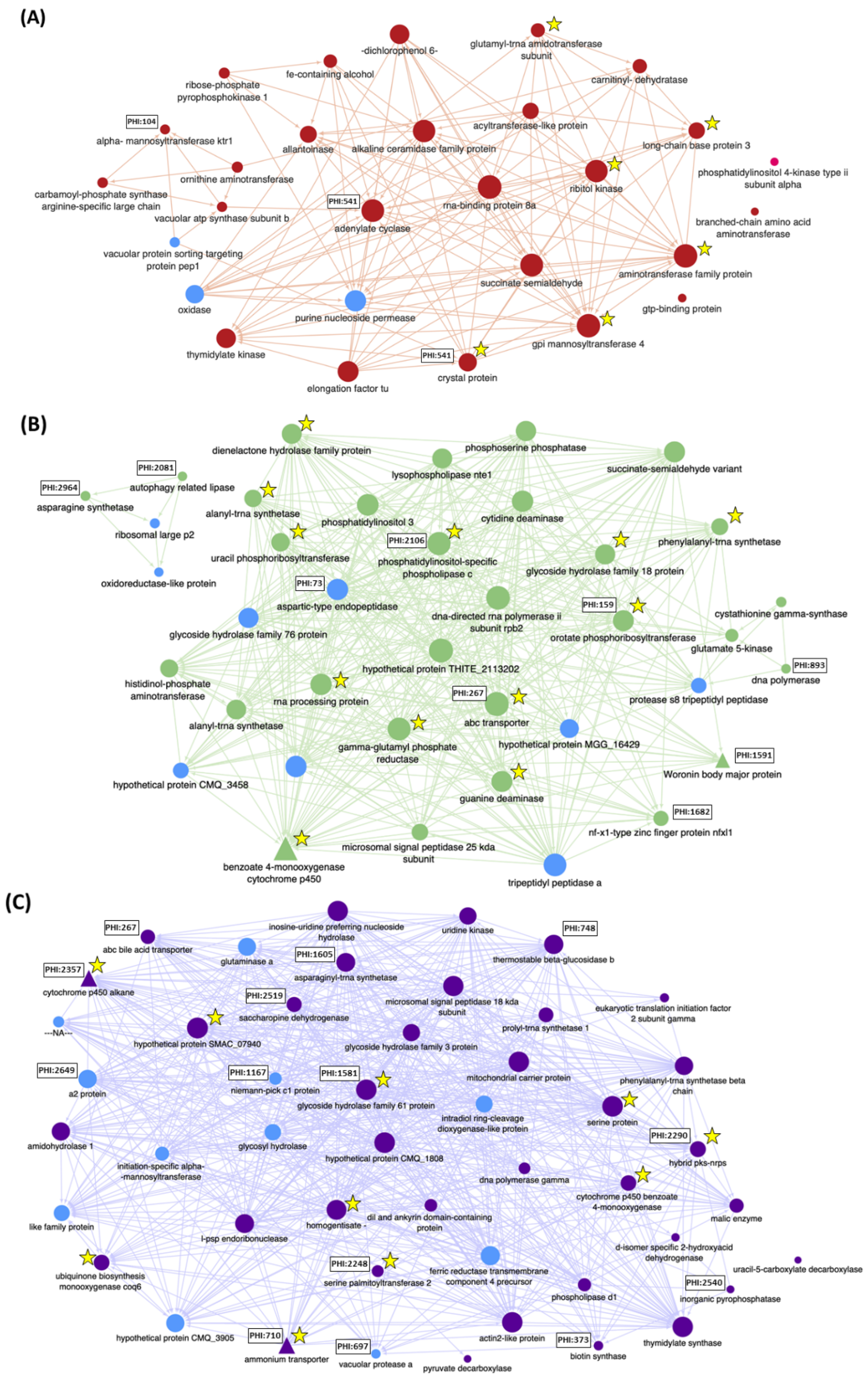
| Strain | Gene | Annotation | PHI: Base | Method 1 | %RGE 2 | Nb Transcripts 3 | DEG 4 | Module 5 | Mycelial Gowth 6 | Virulence 7 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 96_S | 96_VF | MEA | NaCl | Lim | Apple | Elm | ||||||||
| Cyp570-A4D | 7411 | Pisatin demethylase | − | RNAi | 25.3 | 5.0 | 0.0 | Up96_S | Gre | 131.3 8 | n.t. 9 | 81.0 * | 97.0 | 96.3 |
| Cyp52P6-AD | 7466 | Putative α-Pinene to Verbenol enzyme | − | RNAi | 3.4 | 17.3 | 1.0 | Up96_S | Pur | 117.8 | n.t. | 69.0 * | 105.0 | 87.5 |
| Hex1-D | 6790.1 | Hex1 Woronin body formation | 1591 | RNAi | 6.5 | 389.7 | 97.0 | No | Gre | 78.0 * | 45.2 * | 53.7 * | 55.0 * | 100.6 |
| Mad1-J | 3773 | Mad1 Adhesin | − | RNAi | 2.8 | 10.3 | 4.3 | No | Mag | 128.5 * | 145.2 * | 108.9 | 84.0 | 101.0 |
| AmtA-B | 282 | Ammonium transporter | 2710 | RNAi | 1.6 | 13.3 | 0.0 | Up96_S | Pur | 109.2 | n.t. | n.t. | 109.0 | 101.0 |
| ΔAox1 5-1 | 5955 | Aox1 Alcohol oxidase | 199 | OSCAR | n.t. | 567.3 | 808.0 | Up96_VF | GrY | 84.9 * | n.t. | n.t. | 62.2 * | 74.2 |
| ΔOpf2 1-29 | 1642 | Opf2 Transcription Factor | 1931 | OSCAR | n.t. | 33.0 | 16.0 | No | GrY | 100.7 | n.t. | n.t. | 69.4 * | 100.1 |
| ΔBct2 3-12 | 2340 | Bct2 Transcription Factor | 1933 | OSCAR | n.t. | 20.3 | 4.0 | No | Ora | 81.6 * | n.t. | n.t. | 96.9 | 100.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigg, M.; de Oliveira, T.C.; Sarmiento-Villamil, J.L.; de la Bastide, P.Y.; Hintz, W.E.; Sherif, S.M.; Shukla, M.; Bernier, L.; Saxena, P.K. Comparative Analysis of Transcriptomes of Ophiostoma novo-ulmi ssp. americana Colonizing Resistant or Sensitive Genotypes of American Elm. J. Fungi 2022, 8, 637. https://doi.org/10.3390/jof8060637
Nigg M, de Oliveira TC, Sarmiento-Villamil JL, de la Bastide PY, Hintz WE, Sherif SM, Shukla M, Bernier L, Saxena PK. Comparative Analysis of Transcriptomes of Ophiostoma novo-ulmi ssp. americana Colonizing Resistant or Sensitive Genotypes of American Elm. Journal of Fungi. 2022; 8(6):637. https://doi.org/10.3390/jof8060637
Chicago/Turabian StyleNigg, Martha, Thais C. de Oliveira, Jorge L. Sarmiento-Villamil, Paul Y. de la Bastide, Will E. Hintz, Sherif M. Sherif, Mukund Shukla, Louis Bernier, and Praveen K. Saxena. 2022. "Comparative Analysis of Transcriptomes of Ophiostoma novo-ulmi ssp. americana Colonizing Resistant or Sensitive Genotypes of American Elm" Journal of Fungi 8, no. 6: 637. https://doi.org/10.3390/jof8060637