
Population Genomics Reveals Population Structure and Mating-Type Loci in Marssonina brunnea


Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolates Collection
2.2. DNA Extraction and Genome Sequencing
2.3. Read Mapping, SNP Calling, and Summary Statistics
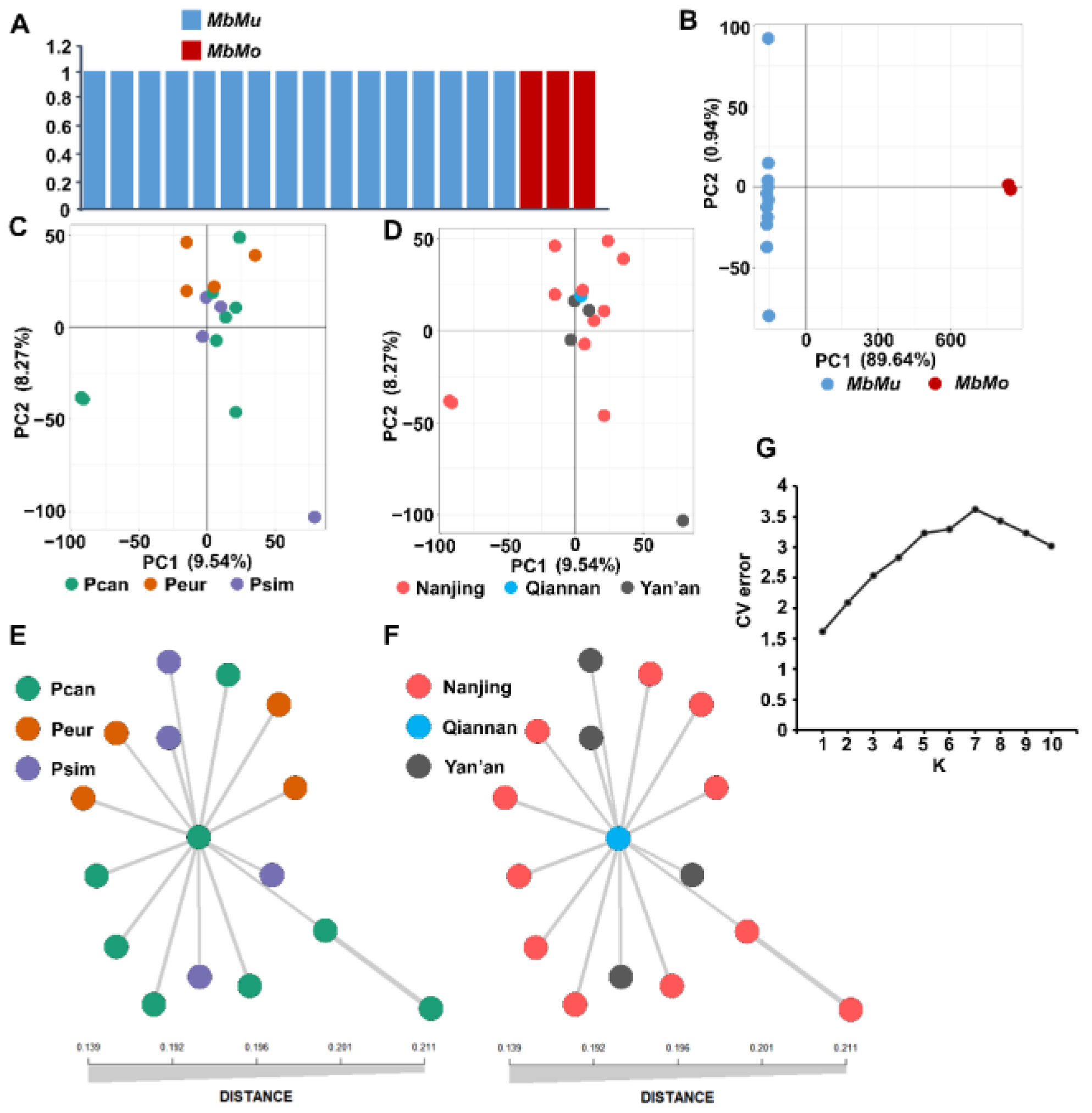
2.4. Population Structure
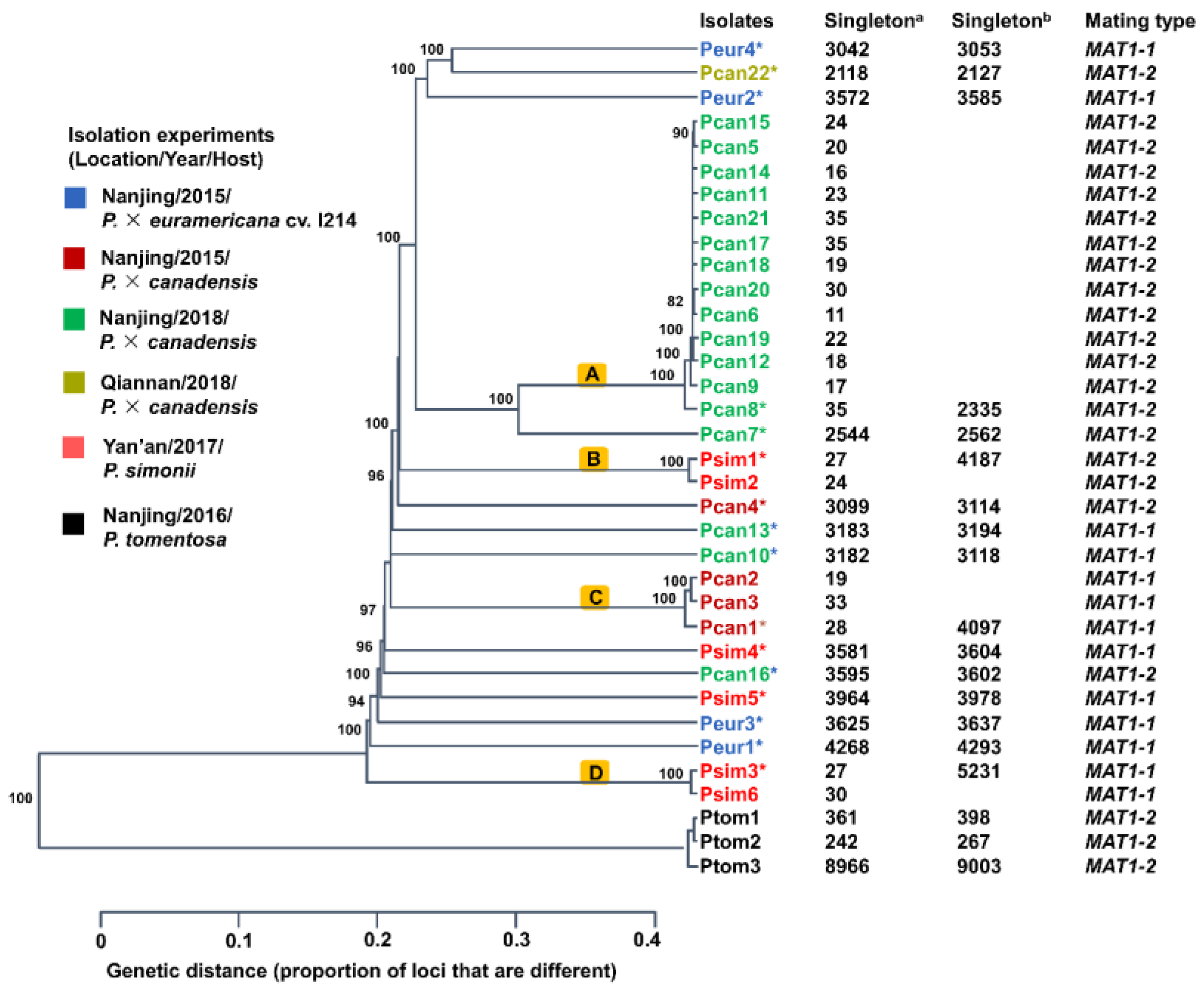
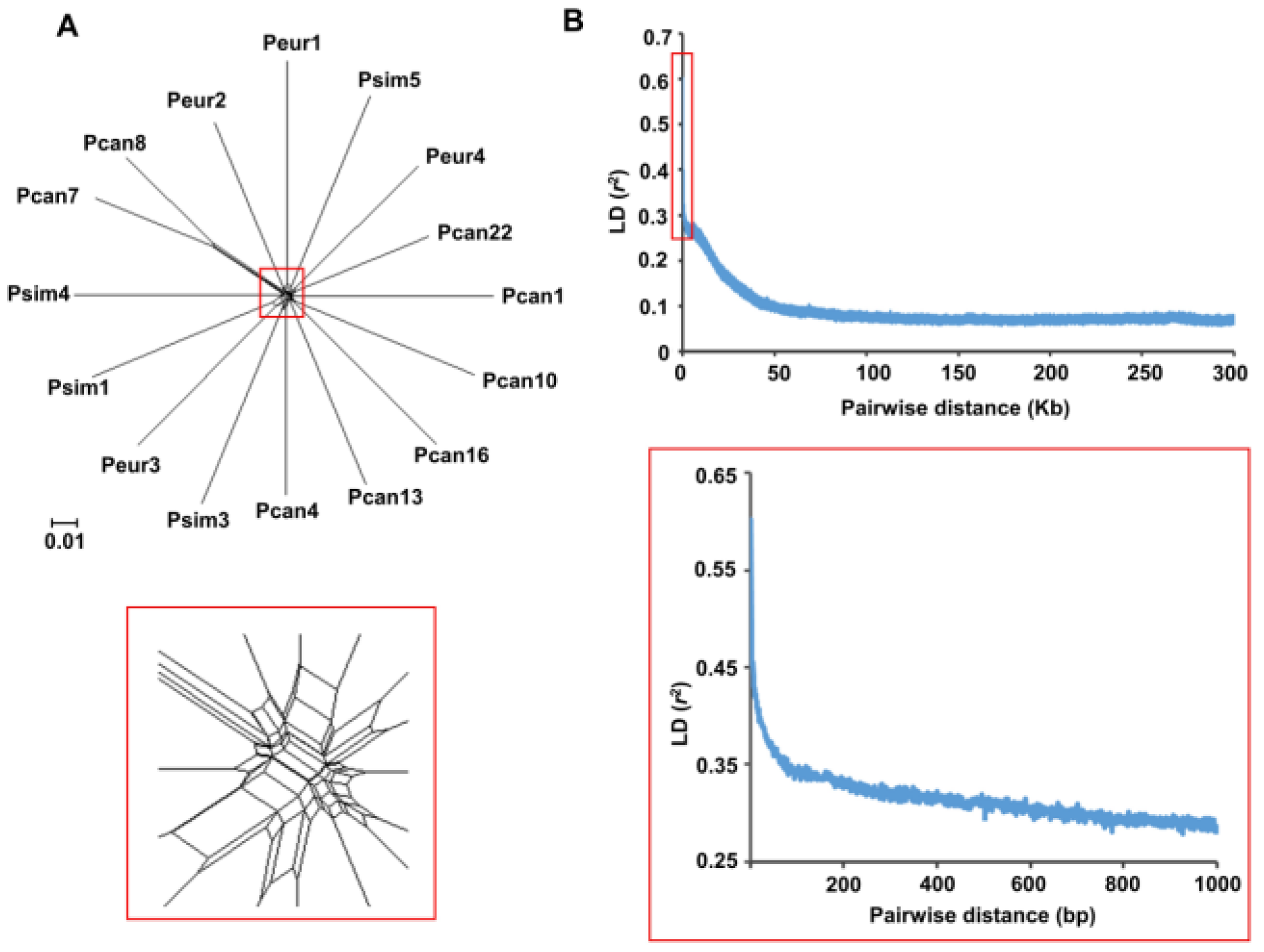
2.5. Phylogeny and Recombination Analysis
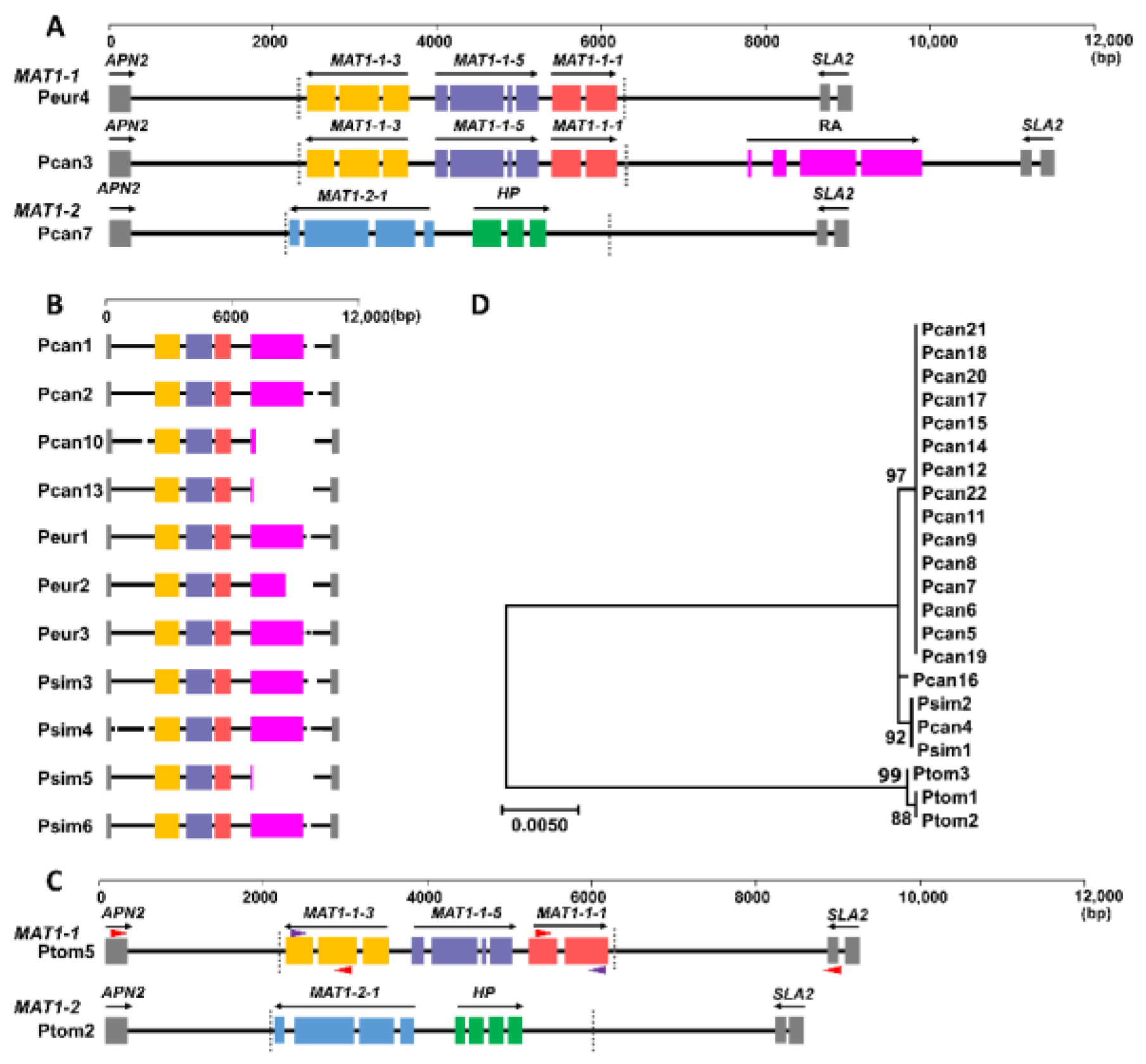
2.6. Genome Assembly and MAT Loci
3. Results
3.1. Resequencing and SNP Calling of MbMu and MbMo
3.2. Population Structure Analysis of MbMu and MbMo
3.3. Recombination in the MbMu Population
3.4. MAT Loci of M. brunnea
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spiers, A.G.; Wenham, H.T. Poplar seed-transmission of Marssonina brunnea. Eur. J. For. Pathol. 1983, 13, 305–314. [Google Scholar] [CrossRef]
- Ellis, J.; Everhart, B. New and rare species of North American fungi (Sphœropsideœ). J. Mycol. 1889, 5, 145–157. [Google Scholar] [CrossRef]
- Gennaro, M.; Giorcelli, A. The biotic adversities of poplar in Italy: A reasoned analysis of factors determining the current state and future perspectives. Ann. Silvic. Res. 2019, 43, 41–51. [Google Scholar]
- Han, Z.M.; Yin, T.M.; Li, C.D.; Huang, M.R.; Wu, R.L. Host effect on genetic variation of Marssonina brunnea pathogenic to poplars. Theor. Appl. Genet. 2000, 100, 614–620. [Google Scholar] [CrossRef]
- Spiers, A.G. Comparative studies of host specificity and symptoms exhibited by poplars infected with Marssonina brunnea, Marssonina castagnei and Marssonina populi. Eur. J. For. Pathol. 1984, 14, 202–218. [Google Scholar] [CrossRef]
- Jiang, H.R.; Wan, Z.B.; Liu, M.; Hou, J.; Yin, T.M. Mapping quantitative trait loci conferring resistance to Marssonina leaf spot disease in Populus deltoides. Trees 2019, 33, 697–706. [Google Scholar] [CrossRef]
- Spiers, A.G.; Hopcroft, D.H. Studies of microconidia of Marssonina brunnea and apothecia of Drepanopeziza tremulae. Eur. J. For. Pathol. 1986, 16, 65–82. [Google Scholar] [CrossRef]
- Wilken, P.M.; Steenkamp, E.T.; Wingfield, M.J.; De Beer, Z.W.; Wingfield, B.D. Which MAT gene? Pezizomycotina (Ascomycota) mating-type gene nomenclature reconsidered. Fungal Biol. Rev. 2017, 31, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Cao, Y.Z.; Jiang, C.; Tan, B.Y.; Wang, Z.; Feng, S.; Zhang, L.; Su, X.H.; Brejova, B.; Vinar, T.; et al. Sequencing the genome of Marssonina brunnea reveals fungus-poplar co-evolution. BMC Genom. 2012, 13, 382. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwenhuis, B.P.; James, T.Y. The frequency of sex in fungi. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, F.E.; Duhamel, M.; Carpentier, F.; Hood, M.E.; Foulongne-Oriol, M.; Silar, P.; Malagnac, F.; Grognet, P.; Giraud, T. Recombination suppression and evolutionary strata around mating-type loci in fungi: Documenting patterns and understanding evolutionary and mechanistic causes. New Phytol. 2021, 229, 2470–2491. [Google Scholar] [CrossRef] [PubMed]
- Abate, D.; De Miccolis Angelini, R.M.; Rotolo, C.; Pollastro, S.; Faretra, F. Mating System in the Brown Rot Pathogens Monilinia fructicola, M. laxa, and M. fructigena. Phytopathology 2018, 108, 1315–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wey, T.; Schlegel, M.; Stroheker, S.; Gross, A. MAT--gene structure and mating behavior of Hymenoscyphus fraxineus and Hymenoscyphus albidus. Fungal Genet. Biol. 2016, 87, 54–63. [Google Scholar] [CrossRef]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [Green Version]
- Chitrampalam, P.; Pryor, B.M. Characterization of mating type (MAT) alleles differentiated by a natural inversion in Sclerotinia minor. Plant Pathol. 2015, 64, 911–920. [Google Scholar] [CrossRef]
- Robicheau, B.M.; Bunbury-Blanchette, A.L.; LaButti, K.; Grigoriev, I.V.; Walker, A.K. The homothallic mating-type locus of the conifer needle endophyte Phialocephala scopiformis DAOMC 229536 (order Helotiales). Fungal Biol. 2017, 121, 1011–1024. [Google Scholar] [CrossRef]
- Penselin, D.; Munsterkotter, M.; Kirsten, S.; Felder, M.; Taudien, S.; Platzer, M.; Ashelford, K.; Paskiewicz, K.H.; Harrison, R.J.; Hughes, D.J.; et al. Comparative genomics to explore phylogenetic relationship, cryptic sexual potential and host specificity of Rhynchosporium species on grasses. BMC Genom. 2016, 17, 953. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Chen, J.X.; Zhao, L.J. Draft genome sequence of Marssonina coronaria, causal agent of apple blotch, and comparisons with the Marssonina brunnea and Marssonina rosae genomes. PLoS ONE 2021, 16, e0246666. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate a | Sampling Experiment b | Clean Data (Gb) | Assembly (Mb) | Genome Coverage | Average Read Depth | SNP c | SNP d | SNP e |
|---|---|---|---|---|---|---|---|---|
| Peur1 * | 1 | 2.36 | 35.52 | 95.51% | 32.97 | 42,314 | 40,633 | 99,752 |
| Peur2 * | 1 | 2.66 | 36.06 | 95.64% | 38.14 | 39,390 | 38,066 | 88,244 |
| Peur3 * | 1 | 3.12 | 35.79 | 96.82% | 44.05 | 42,252 | 40,766 | 93,741 |
| Peur4 * | 1 | 2.28 | 36.01 | 96.37% | 33.48 | 39,157 | 37,698 | 88,639 |
| Pcan1 * | 2 | 2.04 | 33.8 | 92.65% | 32.61 | 42,501 | 40,961 | 94,116 |
| Pcan2 | 2 | 2.79 | 36.18 | 96.58% | 39.06 | 42,438 | ||
| Pcan3 | 2 | 2.34 | 35.06 | 93.59% | 36.15 | 42,455 | ||
| Pcan4 * | 2 | 3.26 | 35.74 | 93.69% | 47.27 | 40,182 | 38,829 | 91,156 |
| Pcan5 | 3 | 2.56 | 36.86 | 93.83% | 40.62 | 38,552 | ||
| Pcan6 | 3 | 2.72 | 35.22 | 92.97% | 41.42 | 38,531 | ||
| Pcan7 * | 3 | 2.03 | 35.34 | 96.24% | 31 | 39,548 | 38,055 | 94,022 |
| Pcan8 * | 3 | 2.15 | 34.23 | 88.99% | 35.86 | 38,622 | 37,171 | 92,645 |
| Pcan9 | 3 | 2.28 | 34.94 | 92.11% | 34.92 | 38,558 | ||
| Pcan10 * | 3 | 2.56 | 36.12 | 96.96% | 38.9 | 40,256 | 38,844 | 90,990 |
| Pcan11 | 3 | 2.00 | 35.18 | 93.17% | 31.2 | 38,551 | ||
| Pcan12 | 3 | 2.64 | 35.78 | 92.91% | 40.05 | 38,563 | ||
| Pcan13 * | 3 | 2.56 | 35.88 | 95.62% | 35.92 | 38,977 | 37,539 | 90,490 |
| Pcan14 | 3 | 2.85 | 35.97 | 93.41% | 42.87 | 38,540 | ||
| Pcan15 | 3 | 3.17 | 36.07 | 93.63% | 43.66 | 38,550 | ||
| Pcan16 * | 3 | 2.83 | 36.5 | 97.55% | 38.35 | 39,902 | 38,405 | 90,985 |
| Pcan17 | 3 | 2.58 | 37.44 | 93.35% | 37.65 | 38,543 | ||
| Pcan18 | 3 | 2.05 | 35.79 | 93.25% | 30.16 | 38,546 | ||
| Pcan19 | 3 | 2.08 | 35.75 | 93.52% | 30.8 | 38,546 | ||
| Pcan20 | 3 | 2.22 | 35.72 | 93.61% | 32.58 | 38,547 | ||
| Pcan21 | 3 | 2.32 | 35.97 | 93.69% | 34.95 | 38,551 | ||
| Pcan22 * | 4 | 2.86 | 36.05 | 96.07% | 39.4 | 34,120 | 32,941 | 80,407 |
| Psim1 * | 5 | 2.70 | 36.33 | 96.63% | 39.52 | 42,374 | 40,857 | 93,843 |
| Psim2 | 5 | 2.43 | 35.01 | 95.63% | 37.35 | 42,376 | ||
| Psim3 * | 5 | 3.01 | 40.99 | 97.87% | 35.45 | 44,692 | 43,147 | 95,753 |
| Psim4 * | 5 | 2.19 | 35.87 | 96.32% | 33.46 | 41,279 | 39,671 | 94,038 |
| Psim5 * | 5 | 2.00 | 35.31 | 92.77% | 31.5 | 40,952 | 39,399 | 94,477 |
| Psim6 | 5 | 2.78 | 36.14 | 93.61% | 39.62 | 44,683 | ||
| Ptom1 * | 6 | 16.18 | 50.87 | 48.25% | 127.28 | 1,023,042 | 1,023,392 | |
| Ptom3 * | 6 | 3.5 | 37.16 | 47.38% | 30.88 | 1,023,009 | 1,023,361 | |
| Ptom2 * | 6 | 3.81 | 37.06 | 47.49% | 31.46 | 1,022,870 | 1,023,223 | |
| Total | 1,139,808 | 1,140,405 | 430,649 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Q.; Yang, H.; Chen, J.; Zhao, L. Population Genomics Reveals Population Structure and Mating-Type Loci in Marssonina brunnea. J. Fungi 2022, 8, 579. https://doi.org/10.3390/jof8060579
Cheng Q, Yang H, Chen J, Zhao L. Population Genomics Reveals Population Structure and Mating-Type Loci in Marssonina brunnea. Journal of Fungi. 2022; 8(6):579. https://doi.org/10.3390/jof8060579
Chicago/Turabian StyleCheng, Qiang, Hougang Yang, Junxiang Chen, and Lijuan Zhao. 2022. "Population Genomics Reveals Population Structure and Mating-Type Loci in Marssonina brunnea" Journal of Fungi 8, no. 6: 579. https://doi.org/10.3390/jof8060579