
Anemochore Seeds Harbor Distinct Fungal and Bacterial Abundance, Composition, and Functional Profiles


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Seed Collection and Surface Sterilization
2.2. Microbial DNA Extraction, PNA Clamps, PCR Amplification, and High-Throughput Sequencing
2.3. Quantitative Real-Time PCR
2.4. Statistical Analysis
3. Results
3.1. Bacterial and Fungal Abundances in Wind- or Water-Dispersed Seeds
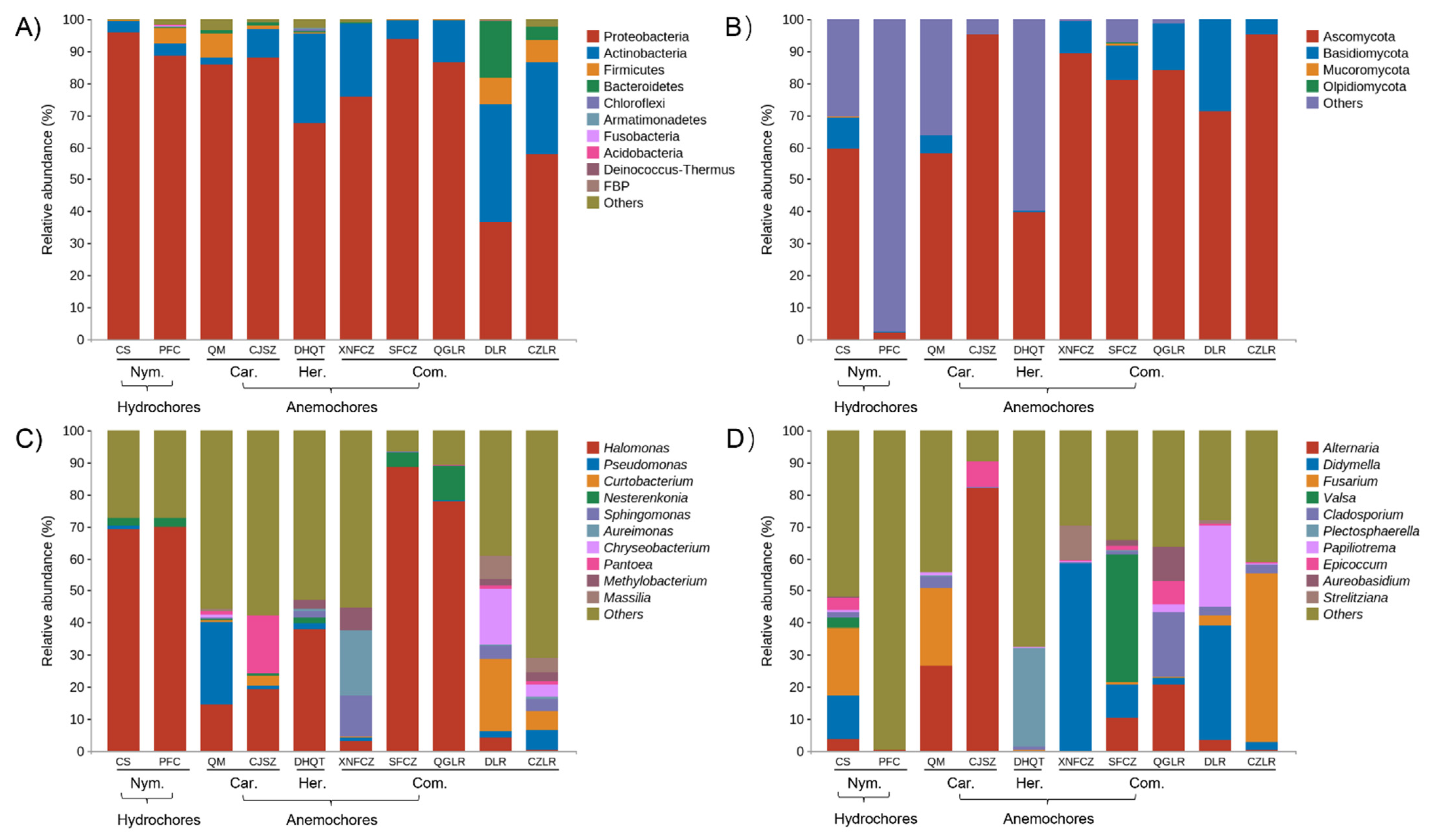
3.2. The Composition of Bacterial and Fungal Taxa within Wind- or Water-Dispersed Seeds
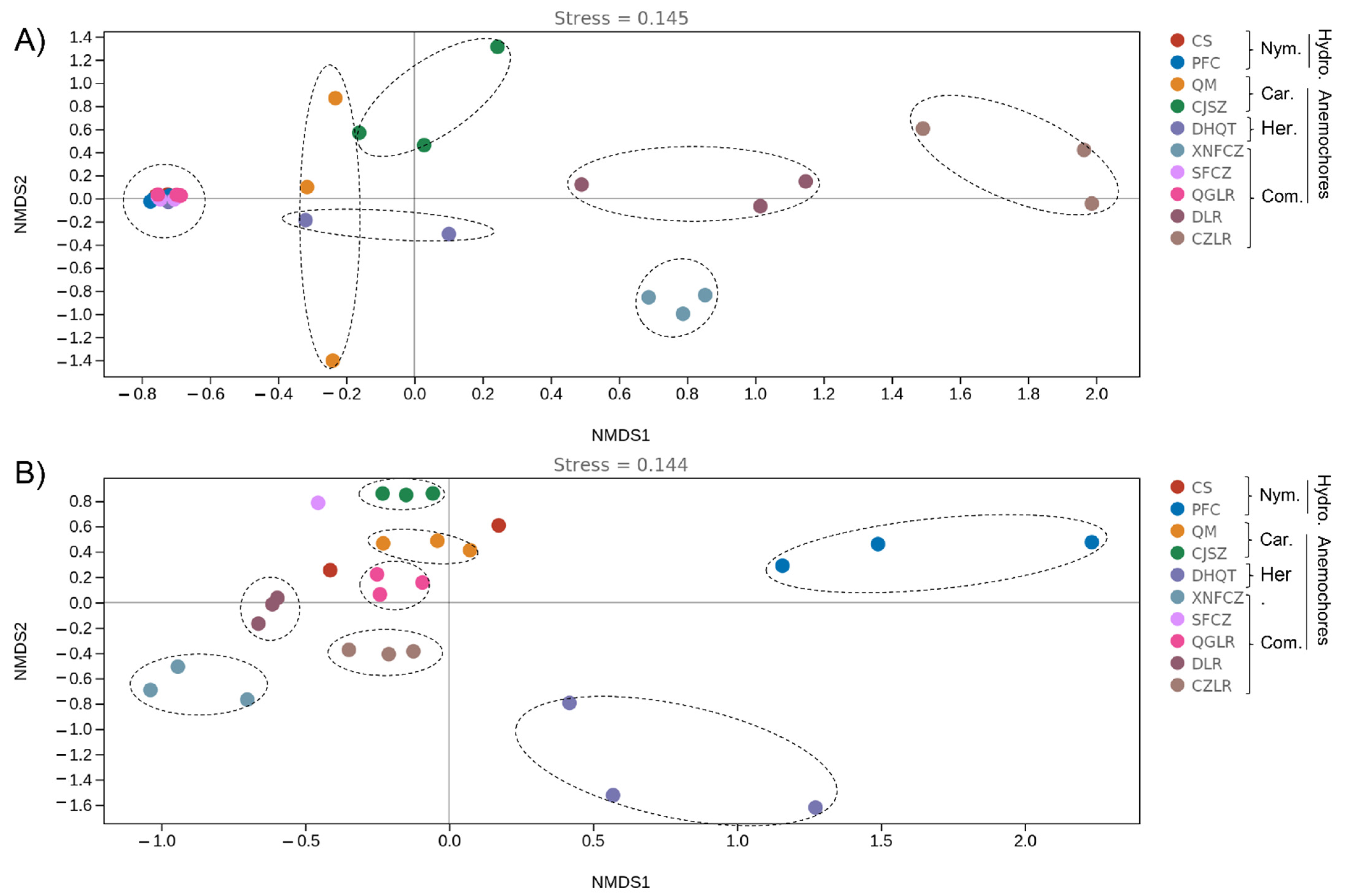
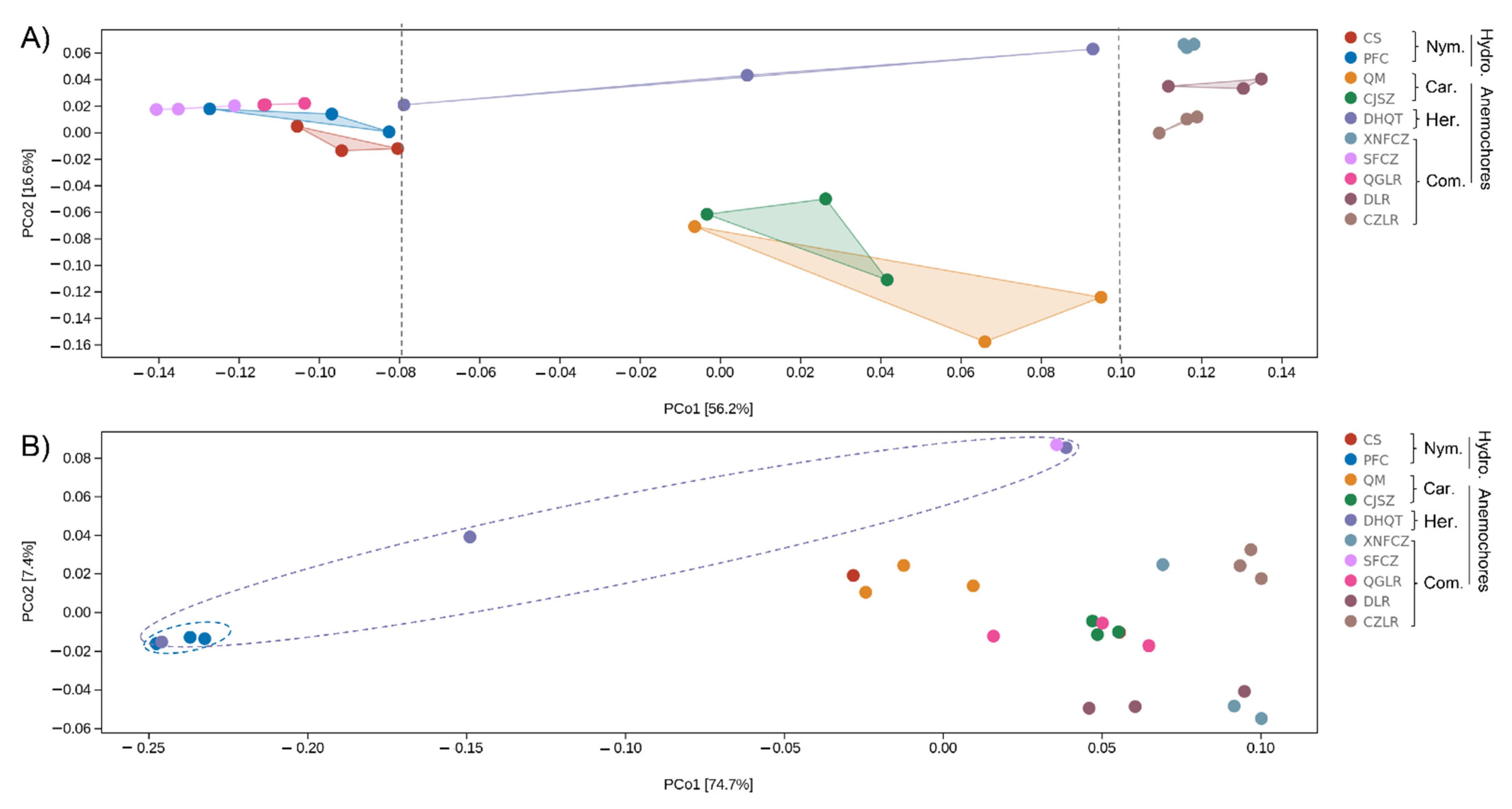
3.3. Identification of the Main Drivers of Microbial Diversity and Structure within Wind- and Water-Dispersed Seeds
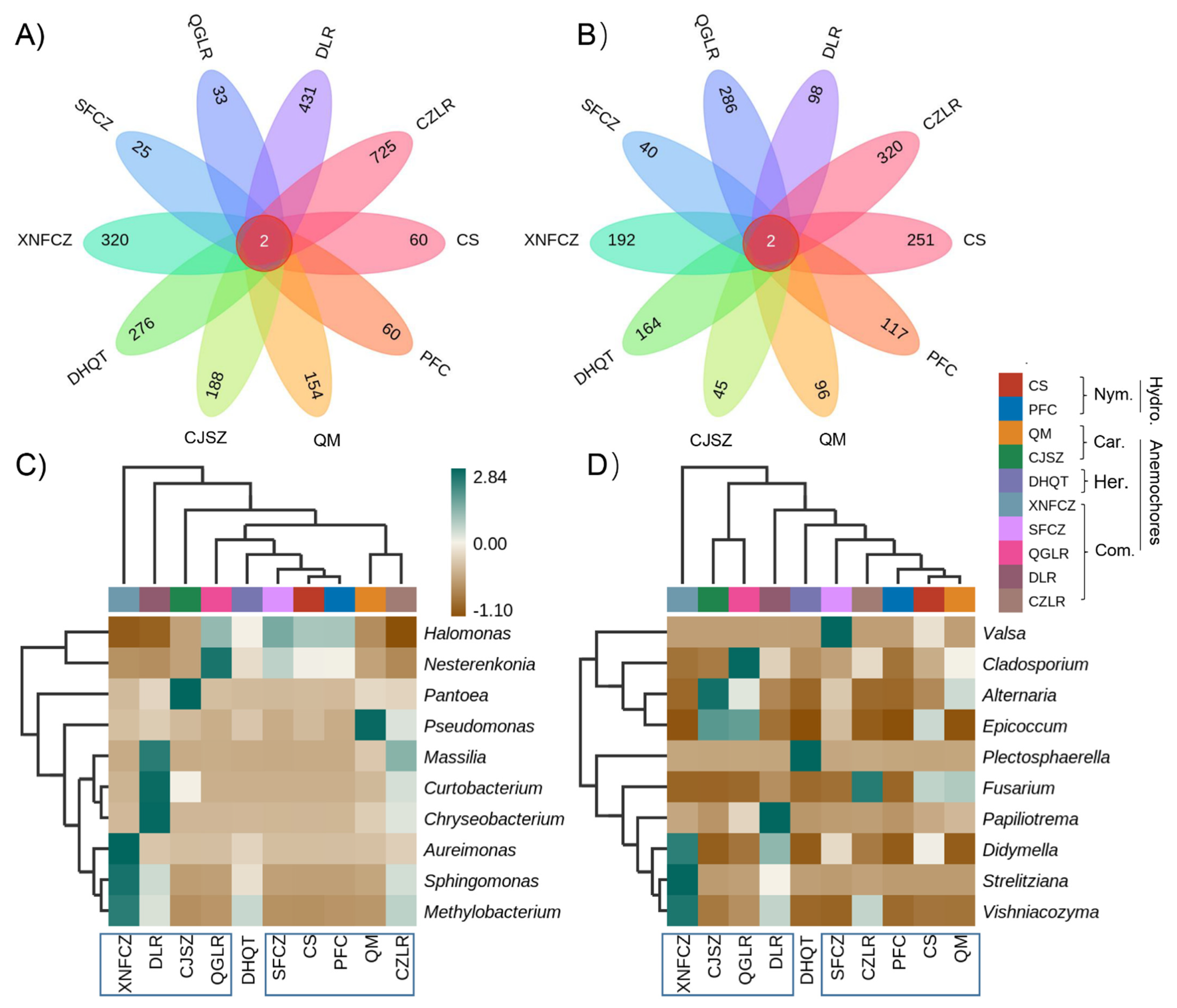
3.4. Species Differences and Marker Species Analysis
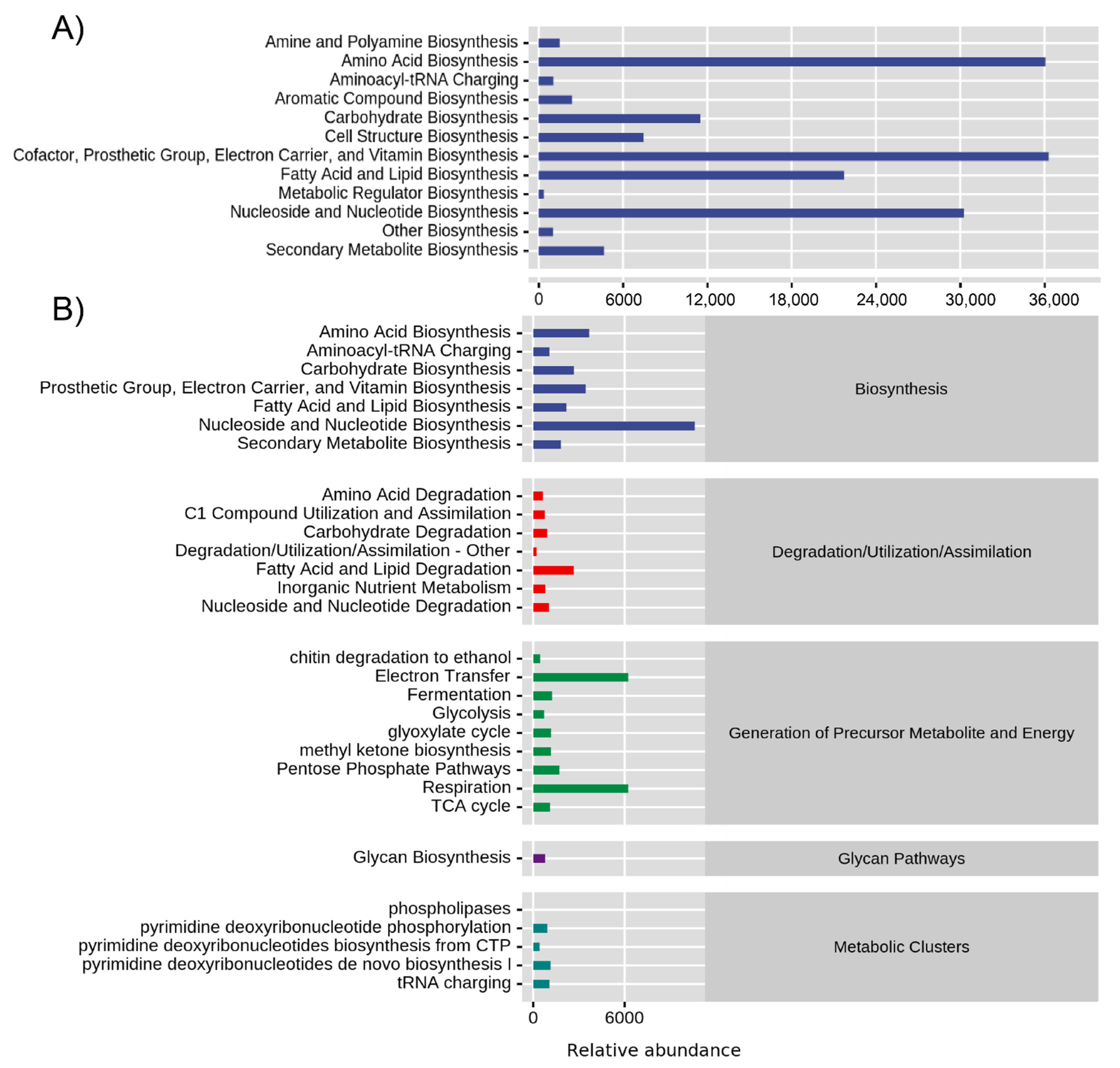
3.5. Microbiome Function
4. Discussion
4.1. Changes in the Seed Microbiome among Anemochores
4.2. Predictive Functional Profiling in the Seed Microbiome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kusstatscher, P.; Adam, E.; Wicaksono, W.A.; Bernhart, M.; Olimi, E.; Müller, H.; Berg, G. Microbiome-assisted breeding to understand cultivar-dependent assembly in Cucurbita pepo. Front. Plant Sci. 2021, 12, 642027. [Google Scholar] [CrossRef]
- Walsh, C.M.; Becker-Uncapher, I.; Carlson, M.; Fierer, N. Variable influences of soil and seed-associated bacterial communities on the assembly of seedling microbiomes. ISME J. 2021, 15, 2748–2762. [Google Scholar] [CrossRef] [PubMed]
- Kuźniar, A.; Włodarczyk, K.; Grządziel, J.; Woźniak, M.; Furtak, K.; Gałązka, A.; Dziadczyk, E.; Skórzyńska-Polit, E.; Wolińska, A. New insight into the composition of wheat seed microbiota. Int. J. Mol. Sci. 2020, 21, 634. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, D.; Mancinelli, R.; Wikström, M.; Birch-Jensen, A.S.; Postma, J.; Ehlers, R.U.; Goertz, S.; Berg, G. The structure of the Brassica napus seed microbiome is cultivar-dependent and affects the interactions of symbionts and pathogens. Microbiome 2017, 5, 104. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, B.; Cernava, T.; Müller, H.; Berg, C.; Berg, G. Seeds of native alpine plants host unique microbial communities embedded in cross-kingdom networks. Microbiome 2019, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wu, H.; Yan, B.; Zhao, H.; Liu, F.; Zhang, H.; Sheng, Q.; Miao, F.; Liang, Z. Core microbiome of medicinal plant salvia miltiorrhiza seed: A rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci. 2018, 19, 672. [Google Scholar] [CrossRef] [Green Version]
- Ismaiel, A.A.; Ahmed, A.S.; Hassan, I.A.; El-Sayed, E.S.R.; Karam El-Din, A.Z.A. Production of paclitaxel with anticancer activity by two local fungal endophytes, Aspergillus fumigatus and Alternaria tenuissima. Appl. Microbiol. Biotechnol. 2017, 101, 5831–5846. [Google Scholar] [CrossRef]
- Soltani, J.; Hosseyni Moghaddam, M.S. Antiproliferative, antifungal, and antibacterial activities of endophytic Alternaria species from Cupressaceae. Curr. Microbiol. 2014, 69, 349–356. [Google Scholar] [CrossRef]
- Rego, C.H.Q.; França-Silva, F.; Gomes-Junior, F.G.; de Moraes, M.H.D.; de Medeiros, A.D.; da Silva, C.B. Using multispectral imaging for detecting seed-borne fungi in cowpea. Agriculture 2020, 10, 361. [Google Scholar] [CrossRef]
- Abdullaeva, Y.; Ambika Manirajan, B.; Honermeier, B.; Schnell, S.; Cardinale, M. Domestication affects the composition, diversity, and co-occurrence of the cereal seed microbiota. J. Adv. Res. 2021, 31, 75–86. [Google Scholar] [CrossRef]
- Shao, J.; Miao, Y.; Liu, K.; Ren, Y.; Xu, Z.; Zhang, N.; Feng, H.; Shen, Q.; Zhang, R.; Xun, W. Rhizosphere microbiome assembly involves seed-borne bacteria in compensatory phosphate solubilization. Soil Biol. Biochem. 2021, 159, 108273. [Google Scholar] [CrossRef]
- Gundel, P.E.; Rudgers, J.A.; Ghersa, C.M. Incorporating the process of vertical transmission into understanding of host-symbiont dynamics. Oikos 2011, 120, 1121–1128. [Google Scholar] [CrossRef]
- Johnston-Monje, D.; Raizada, M.N. Conservation and diversity of seed associated endophytes in Zea across boundaries of evolution, ethnography and ecology. PLoS ONE 2011, 6, e20396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacilio-Jiménez, M.; Aguilar-Flores, S.; Del Valle, M.V.; Pérez, A.; Zepeda, A.; Zenteno, E. Endophytic bacteria in rice seeds inhibit early colonization of roots by Azospirillum brasilense. Soil Biol. Biochem. 2001, 33, 167–172. [Google Scholar] [CrossRef]
- Cottyn, B.; Regalado, E.; Lanoot, B.; De Cleene, M.; Mew, T.W.; Swings, J. Bacterial populations associated with rice seed in the tropical environment. Phytopathology 2001, 91, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Oehrle, N.W.; Karr, D.B.; Kremer, R.J.; Emerich, D.W. Enhanced attachment of Bradyrhizobium japonicom to soybean through reduced root colonization of internally seedborne microorganisms. Can. J. Microbiol. 2000, 46, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Llewellyn, T.; Downes, E.; Oddy, J.; MacIntosh, C.; Kallow, S.; Panis, B.; Dickie, J.B.; Gaya, E. Seed banks as incidental fungi banks: Fungal endophyte diversity in stored seeds of banana wild relatives. Front. Microbiol. 2021, 12, 643731. [Google Scholar] [CrossRef]
- Burke, C.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. USA 2011, 108, 14288–14293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, J.; Huang, L.; He, Z.; Chen, L.; Hua, Z.; Jia, P.; Li, S.; Liu, J.; Li, J.; Zhou, J.; et al. Predicting taxonomic and functional structure of microbial communities in acid mine drainage. ISME J. 2016, 10, 1527–1539. [Google Scholar] [CrossRef]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; Van Veen, J.A.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Jacques, M.A.; Barret, M. Ecological patterns of seed microbiome diversity, transmission, and assembly. Curr. Opin. Microbiol. 2017, 37, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Charles-dominique, P. Relationship between seed dispersal and behavioural ecology. In Dynamics and Plant-Animal Interactions in a Neotropical Rainforest; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; pp. 191–196. [Google Scholar]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Herren, C.M.; McMahon, K.D. Keystone taxa predict compositional change in microbial communities. Environ. Microbiol. 2018, 20, 2207–2217. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Liu, D.; Chater, C.C.C.; Yu, F.; Perez-Moreno, J. Tuber pseudohimalayense ascomata-compartments strongly select their associated bacterial microbiome from nearby pine forest soils independently of their maturation stage. Pedobiologia 2021, 87–88, 150743. [Google Scholar] [CrossRef]
- Arnold, A.E.; Henk, D.A.; Eells, R.L.; Lutzoni, F.; Vilgalys, R. Diversity and phylogenetic affinities of foliar fungal endophytes in loblolly pine inferred by culturing and environmental PCR. Mycologia 2007, 99, 185–206. [Google Scholar] [CrossRef]
- Horton, M.W.; Bodenhausen, N.; Beilsmith, K.; Meng, D.; Muegge, B.D.; Subramanian, S.; Vetter, M.M.; Vilhjálmsson, B.J.; Nordborg, M.; Gordon, J.I.; et al. Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Orgiazzi, A.; Lumini, E.; Nilsson, R.H.; Girlanda, M.; Vizzini, A.; Bonfante, P.; Bianciotto, V. Unravelling soil fungal communities from different mediterranean land-use backgrounds. PLoS ONE 2012, 7, e34847. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef]
- Yu, X.; Zhu, Y.; Wang, B.; Liu, D.; Bai, H.; Jin, L.; Wang, B.; Ruan, H.; Mao, L.; Jin, F.; et al. Effects of nitrogen addition on rhizospheric soil microbial communities of poplar plantations at different ages. For. Ecol. Manag. 2021, 494, 119328. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; He, X.; Chater, C.C.C.; Perez-Moreno, J.; Yu, F. Microbiome community structure and functional gene partitioning in different micro-niches within a sporocarp-forming fungus. Front. Microbiol. 2021, 12, 629352. [Google Scholar] [CrossRef]
- Chen, Q.L.; Ding, J.; Zhu, Y.G.; He, J.Z.; Hu, H.W. Soil bacterial taxonomic diversity is critical to maintaining the plant productivity. Environ. Int. 2020, 140, 105766. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Kahl, G. UPGMA. In The Dictionary of Genomics, Transcriptomics and Proteomics; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; p. 1. [Google Scholar]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2008, 42, D459–D471. [Google Scholar] [CrossRef] [Green Version]
- Truyens, S.; Weyens, N.; Cuypers, A.; Vangronsveld, J. Bacterial seed endophytes: Genera, vertical transmission and interaction with plants. Environ. Microbiol. Rep. 2015, 7, 40–50. [Google Scholar] [CrossRef]
- Rijavec, T.; Lapanje, A.; Dermastia, M.; Rupnik, M. Isolation of bacterial endophytes from germinated maize kernels. Can. J. Microbiol. 2007, 53, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, P.D.; Wehmeyer, U.; Stackebrandt, E. Halomonadaceae fam. nov., a new family of the class proteobacteria to accommodate the genera Halomonas and Deleya. Syst. Appl. Microbiol. 1988, 11, 16–19. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Tsuge, T.; Harimoto, Y.; Akimitsu, K.; Ohtani, K.; Kodama, M.; Akagi, Y.; Egusa, M.; Yamamoto, M.; Otani, H. Host-selective toxins produced by the plant pathogenic fungus Alternaria alternata. FEMS Microbiol. Rev. 2013, 37, 44–66. [Google Scholar] [CrossRef]
- Simonin, M.; Briand, M.; Chesneau, G.; Rochefort, A.; Marais, C.; Sarniguet, A.; Barret, M. Seed microbiota revealed by a large-scale meta-analysis including 50 plant species. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bensch, K.; Braun, U.; Groenewald, J.Z.; Crous, P.W. The genus cladosporium. Stud. Mycol. 2012, 72, 1–401. [Google Scholar] [CrossRef] [Green Version]
- Thomma, B.P.H.J. Alternaria spp.: From general saprophyte to specific parasite. Mol. Plant Pathol. 2003, 4, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Frossard, A.; Gerull, L.; Mutz, M.; Gessner, M.O. Disconnect of microbial structure and function: Enzyme activities and bacterial communities in nascent stream corridors. ISME J. 2012, 6, 680–691. [Google Scholar] [CrossRef] [Green Version]
- Purahong, W.; Schloter, M.; Pecyna, M.J.; Kapturska, D.; Däumlich, V.; Mital, S.; Buscot, F.; Hofrichter, M.; Gutknecht, J.L.M.; Krüger, D. Uncoupling of microbial community structure and function in decomposing litter across beech forest ecosystems in Central Europe. Sci. Rep. 2014, 4, 7014. [Google Scholar] [CrossRef] [Green Version]
- Guimarães, R.A.; Pherez-Perrony, P.E.; Müller, H.; Berg, G.; Medeiros, F.H.V.; Cernava, T. Microbiome-guided evaluation of Bacillus subtilis BIOUFLA2 application to reduce mycotoxins in maize kernels. Biol. Control 2020, 150, 104370. [Google Scholar] [CrossRef]
- Passera, A.; Follador, A.; Morandi, S.; Miotti, N.; Ghidoli, M.; Venturini, G.; Quaglino, F.; Brasca, M.; Casati, P.; Pilu, R.; et al. Bacterial communities in the embryo of maize landraces: Relation with susceptibility to Fusarium Ear Rot. Microorganisms 2021, 9, 2388. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CS | PFC | QM | CJSZ | DHQT | XNFCZ | SFCZ | QGLR | DLR | CZLR | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bacteria | Mean | 3.5 D | 3.2 D | 5.0 C | 6.0 B | 5.2 C | 6.1 B | 2.2 E | 2.6 D | 6.6 B | 7.9 A |
| SD | 0.3 | 0.4 | 0.7 | 0.2 | 1.0 | 0.0 | 0.2 | 0.2 | 0.4 | 0.2 | |
| Fungi | Mean | 4.8 | 1.0 C | 3.3 B | 1.2 C | 2.4 B | 2.6 B | 4.2 | 4.6 A | 3.3 B | 5.0 A |
| SD | NA | 0.2 | 0.2 | 0.4 | 0.9 | 1.5 | NA | 0.1 | 0.1 | 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Cai, J.; He, H.; Yang, S.; Chater, C.C.C.; Yu, F. Anemochore Seeds Harbor Distinct Fungal and Bacterial Abundance, Composition, and Functional Profiles. J. Fungi 2022, 8, 89. https://doi.org/10.3390/jof8010089
Liu D, Cai J, He H, Yang S, Chater CCC, Yu F. Anemochore Seeds Harbor Distinct Fungal and Bacterial Abundance, Composition, and Functional Profiles. Journal of Fungi. 2022; 8(1):89. https://doi.org/10.3390/jof8010089
Chicago/Turabian StyleLiu, Dong, Jie Cai, Huajie He, Shimei Yang, Caspar C. C. Chater, and Fuqiang Yu. 2022. "Anemochore Seeds Harbor Distinct Fungal and Bacterial Abundance, Composition, and Functional Profiles" Journal of Fungi 8, no. 1: 89. https://doi.org/10.3390/jof8010089