
Comprehensive Assessment of the Virulence Factors sub 3, sub 6 and mcpA in the Zoonotic Dermatophyte Trichophyton benhamiae Using FISH and qPCR


Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Isolates, GPSE Culture and Infection Experiments
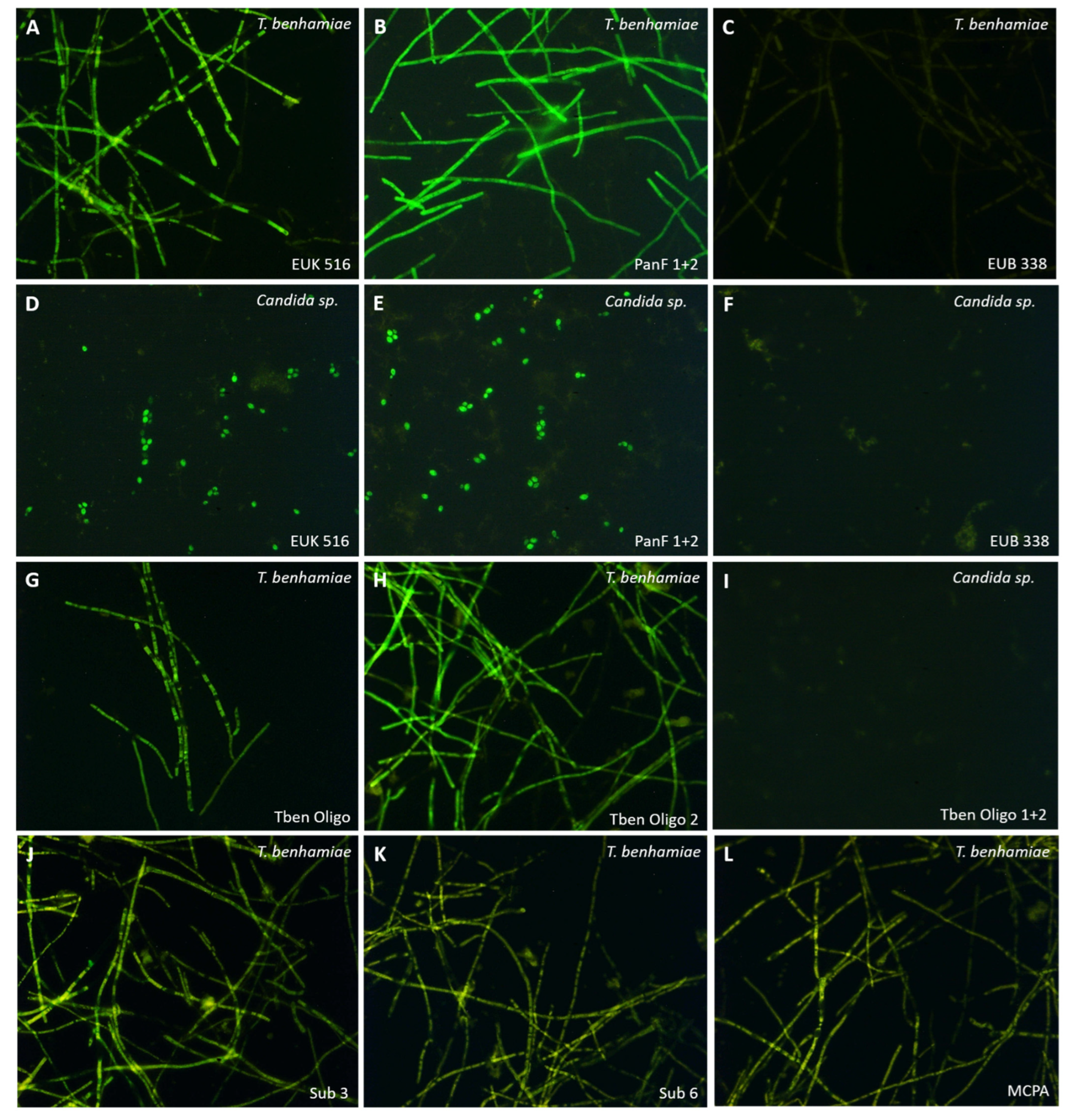
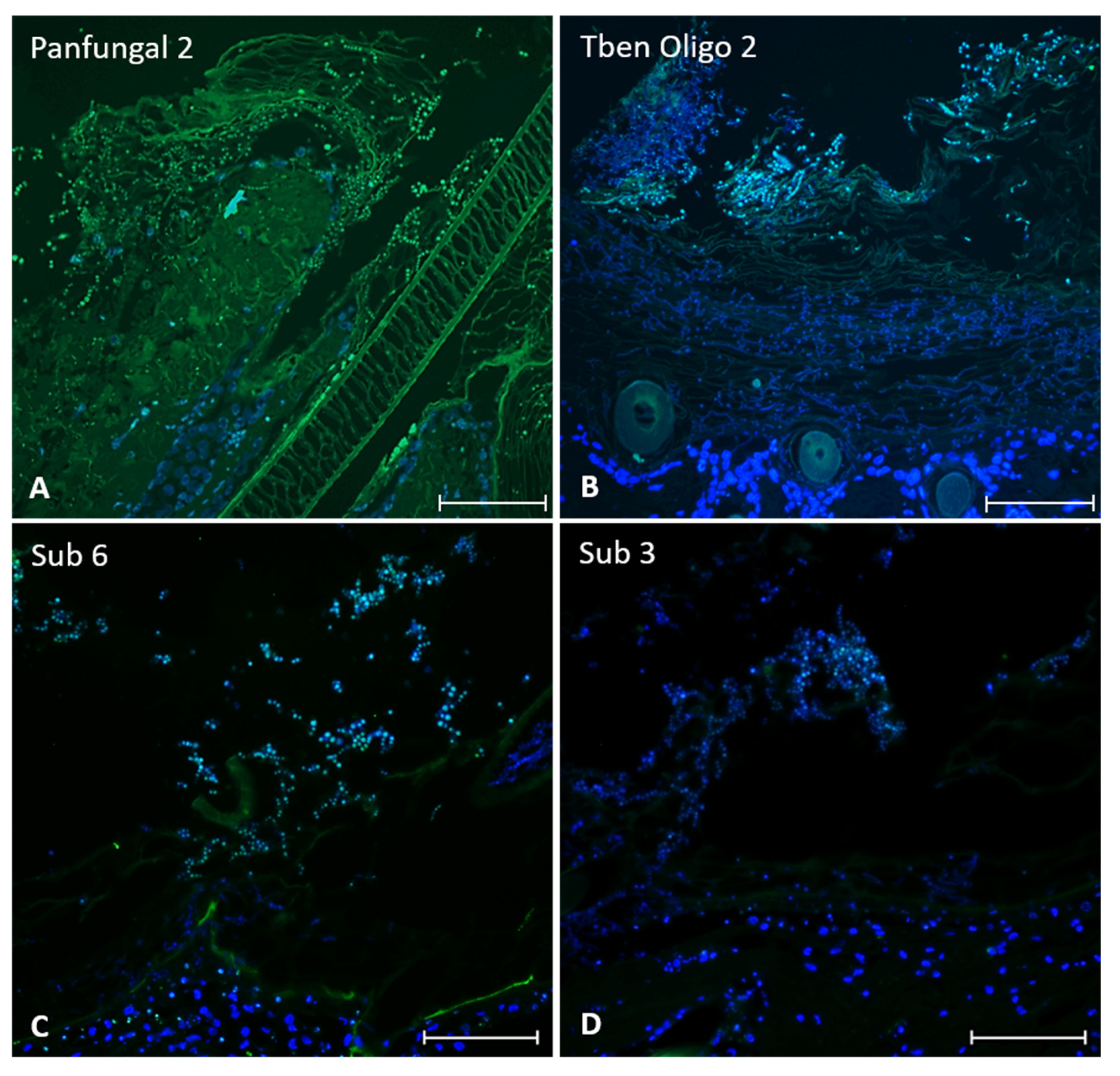
2.2. FISH
2.3. RNA Isolation and qPCR
2.4. Statistics
3. Results
3.1. FISH
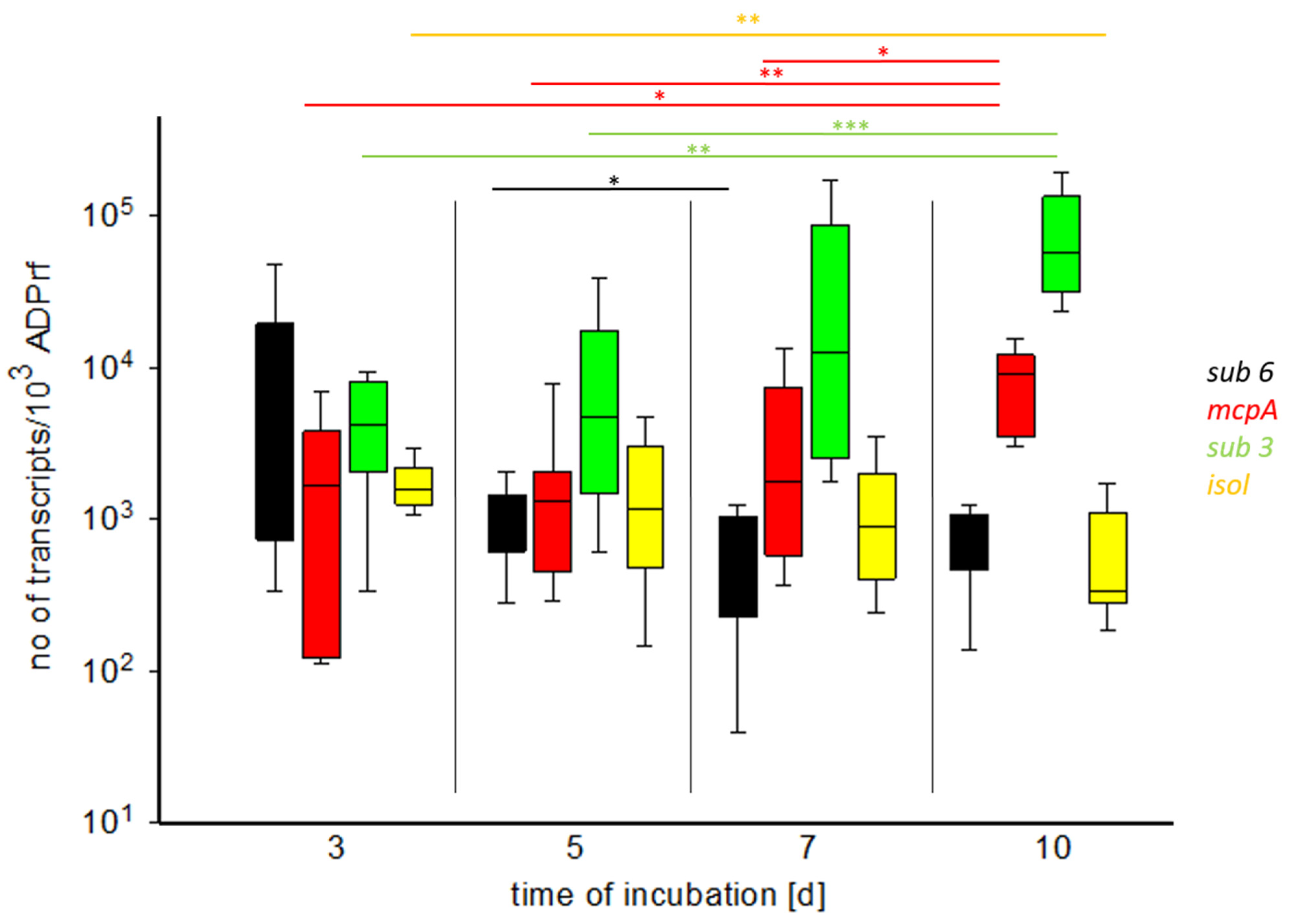
3.2. RNA Isolation and qPCR
4. Discussion
4.1. FISH
4.2. qPCR of Virulence Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Havlickova, B.; Czaika, V.A.; Friedrich, M. Epidemiological trends in skin mycoses worldwide. Mycoses 2008, 51, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Saunte, D.M.L.; Hare, R.K.; Jørgensen, K.M.; Jørgensen, R.; Deleuran, M.; Zachariae, C.O.; Thomsen, S.F.; Bjørnskov-Halkier, L.; Kofoed, K.; Arendrup, M.C. Emerging Terbinafine Resistance in Trichophyton: Clinical Characteristics, Squalene Epoxidase Gene Mutations, and a Reliable EUCAST Method for Detection. Antimicrob. Agents Chemother. 2019, 63, e01126-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, A.; Monod, M.; Salamin, K.; Burmester, A.; Uhrlaß, S.; Wiegand, C.; Hipler, U.-C.; Krüger, C.; Koch, D.; Wittig, F.; et al. Alarming India-wide phenomenon of antifungal resistance in dermatophytes: A multicentre study. Mycoses 2020, 63, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Achterman, R.R.; White, T.C. Dermatophyte Virulence Factors: Identifying and Analyzing Genes That May Contribute to Chronic or Acute Skin Infections. Int. J. Microbiol. 2012, 2012, 358305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staib, P.; Zaugg, C.; Mignon, B.; Weber, J.; Grumbt, M.; Pradervand, S.; Harshman, K.; Monod, M. Differential gene expression in the pathogenic dermatophyte Arthroderma benhamiae in vitro versus during infection. Microbiology 2010, 156, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Tran, V.D.T.; De Coi, N.; Feuermann, M.; Schmid-Siegert, E.; Băguţ, E.-T.; Mignon, B.; Waridel, P.; Peter, C.; Pradervand, S.; Pagni, M.; et al. RNA Sequencing-Based Genome Reannotation of the Dermatophyte Arthroderma benhamiae and Characterization of Its Secretome and Whole Gene Expression Profile during Infection. mSystems 2016, 1, e00036-16. [Google Scholar] [CrossRef] [Green Version]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Methuen: London, UK, 1959. [Google Scholar]
- Quatrin, M.P.; Flores Dalla Lana, D.; Andrzejewski Kaminski, T.F.; Meneghello Fuentefria, A. Fungal infection models: Current progress of ex vivo methods. Mycoses 2019, 62, 860–873. [Google Scholar] [CrossRef]
- Baumbach, C.; Schrödl, W.; Nenoff, P.; Uhrlaß, S.; Mülling, C.K.W.; Michler, J.K. Modeling dermatophytosis: Guinea pig skin explants represent a highly suitable model to study Trichophyton benhamiae infections. J. Dermatol. 2020, 47, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Baumbach, C.; Michler, J.K.; Nenoff, P.; Uhrlaß, S.; Schrödl, W. Visualising virulence factors: Trichophyton benhamiaes subtilisins demonstrated in a guinea pig skin ex vivo model. Mycoses 2020, 63, 970–978. [Google Scholar] [CrossRef]
- Monod, M. Secreted Proteases from Dermatophytes. Mycopathologia 2008, 166, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Leng, W.; Liu, T.; Wang, J.; Li, R.; Jin, Q. Expression dynamics of secreted protease genes in Trichophyton rubrum induced by key host’s proteinaceous components. Med. Mycol. 2009, 47, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldo, A.; Mathy, A.; Tabart, J.; Camponova, P.; Vermout, S.; Massart, L.; Maréchal, F.; Galleni, M.; Mignon, B. Secreted subtilisin Sub3 from Microsporum canis is required for adherence to but not for invasion of the epidermis. Br. J. Dermatol. 2010, 162, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Băguţ, E.T.; Baldo, A.; Mathy, A.; Cambier, L.; Antoine, N.; Cozma, V.; Mignon, B. Subtilisin Sub3 is involved in adherence of Microsporum canis to human and animal epidermis. Vet. Microbiol. 2012, 160, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Méhul, B.; De Coi, N.; Grundt, P.; Genette, A.; Voegel, J.J.; Monod, M. Detection of Trichophyton rubrum and Trichophyton interdigitale in onychomycosis using monoclonal antibodies against Sub6 (Tri r 2). Mycoses 2019, 62, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, R.; Fuchs, B.M. Single-cell identification in microbial communities by improved fluorescence in situ hybridization techniques. Nat. Rev. Genet. 2008, 6, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Kempf, V.A.J.; Trebesius, K.; Autenrieth, I.B. Fluorescent In Situ Hybridization Allows Rapid Identification of Microorganisms in Blood Cultures. J. Clin. Microbiol. 2000, 38, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Martins, M.L.; Ferreira, A.; Sampaio, A.; Vieira, R.; Inácio, J. Direct and specific identification of Cryptococcus neoformans in biological samples using fluorescently labelled DNA probes. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Hayden, R.T.; Isotalo, P.A.; Parrett, T.; Wolk, D.M.; Qian, X.; Roberts, G.D.; Lloyd, R.V. In Situ Hybridization for the Differentiation of Aspergillus, Fusarium, and Pseudallescheria Species in Tissue Section. Diagn. Mol. Pathol. 2003, 12, 21–26. [Google Scholar] [CrossRef]
- Montone, K.T.; Livolsi, V.A.; Lanza, D.C.; Kennedy, D.W.; Palmer, J.; Chiu, A.; Feldman, M.D.; Loevner, L.A.; Nachamkin, D.I. In Situ Hybridization for Specific Fungal Organisms in Acute Invasive Fungal Rhinosinusitis. Am. J. Clin. Pathol. 2011, 135, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Rickerts, V.; Smith, I.M.; Mousset, S.; Kommedal, O.; Fredricks, D.N. Deciphering the aetiology of a mixed fungal infection by broad-range PCR with sequencing and fluorescence in situ hybridisation. Mycoses 2013, 56, 681–686. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Svec, D.; Tichopad, A.; Novosadova, V.; Pfaffl, M.W.; Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomol. Detect. Quantif. 2015, 3, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Descamps, F.; Brouta, F.; Baar, D.; Losson, B.; Mignon, B.; Monod, M.; Zaugg, C. Isolation of a Microsporum canis Gene Family Encoding Three Subtilisin-Like Proteases Expressed in vivo. J. Investig. Dermatol. 2002, 119, 830–835. [Google Scholar] [CrossRef] [Green Version]
- Brouta, F.; Descamps, F.; Monod, M.; Vermout, S.; Losson, B.; Mignon, B. Secreted Metalloprotease Gene Family of Microsporum canis. Infect. Immun. 2002, 70, 5676–5683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Rajak, R.C.; Pandey, A.K.; Gräser, Y. Internal Transcribed Spacer (ITS) of rDNA of appendaged and non-appendaged strains of Microsporum gypseum reveals Microsporum appendiculatum as its synonym. Antonie Leeuwenhoek 2006, 89, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Symoens, F.; Jousson, O.; Packeu, A.; Fratti, M.; Staib, P.; Mignon, B.; Monod, M. The dermatophyte species Arthroderma benhamiae: Intraspecies variability and mating behaviour. J. Med. Microbiol. 2013, 62, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.I.; Binder, B.J.; Olson, R.J.; Chisholm, S.W.; Devereux, R.; Stahl, D.A. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 1990, 56, 1919–1925. [Google Scholar] [CrossRef] [Green Version]
- Rickerts, V.; Khot, P.D.; Myerson, D.; Ko, D.L.; Lambrecht, E.; Fredricks, D.N. Comparison of quantitative real time PCR with Sequencing and ribosomal RNA-FISH for the identification of fungi in Formalin fixed, paraffin-embedded tissue specimens. BMC Infect. Dis. 2011, 11, 202. [Google Scholar] [CrossRef] [Green Version]
- Zaugg, C.; Monod, M.; Weber, J.; Harshman, K.; Pradervand, S.; Thomas, J.; Bueno, M.; Giddey, K.; Staib, P. Gene Expression Profiling in the Human Pathogenic Dermatophyte Trichophyton rubrum during Growth on Proteins. Eukaryot. Cell 2009, 8, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Dunn, M.; Ramírez-Trujillo, J.A.; Hernández-Lucas, I. Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology 2009, 155, 3166–3175. [Google Scholar] [CrossRef] [Green Version]
- Young, A.P.; Jackson, D.J.; Wyeth, R.C. A technical review and guide to RNA fluorescence in situ hybridization. PeerJ 2020, 8, e8806. [Google Scholar] [CrossRef] [Green Version]
- Worek, M.; Kwiatkowska, A.; Ciesielska, A.; Jaworski, A.; Kaplan, J.; Miedziak, B.; Deręgowska, A.; Lewinska, A.; Wnuk, M. Identification of dermatophyte species using genomic in situ hybridization (GISH). J. Microbiol. Methods 2014, 100, 32–41. [Google Scholar] [CrossRef]
- Rickerts, V. Identification of fungal pathogens in Formalin-fixed, Paraffin-embedded tissue samples by molecular methods. Fungal Biol. 2016, 120, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Montone, K.T. Differentiation of Fusarium From Aspergillus Species by Colorimetric In Situ Hybridization in Formalin-Fixed, Paraffin-Embedded Tissue Sections Using Dual Fluorogenic-Labeled LNA Probes. Am. J. Clin. Pathol. 2009, 132, 866–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houseley, J.; Tollervey, D. The Many Pathways of RNA Degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doma, M.K.; Parker, R. RNA Quality Control in Eukaryotes. Cell 2007, 131, 660–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharf, S.; Bartels, A.; Kondakci, M.; Pfeffer, K.; Henrich, B.; Haas, R. Introduction of a bead beating step improves fungal DNA extraction from selected patient specimens. Int. J. Med. Microbiol. 2020, 310, 151443. [Google Scholar] [CrossRef]
- Rodrigues, P.; Venâncio, A.; Lima, N. Toxic reagents and expensive equipment: Are they really necessary for the extraction of good quality fungal DNA? Lett. Appl. Microbiol. 2018, 66, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Pernthaler, A.; Amann, R. Simultaneous Fluorescence In Situ Hybridization of mRNA and rRNA in Environmental Bacteria. Appl. Environ. Microbiol. 2004, 70, 5426–5433. [Google Scholar] [CrossRef] [Green Version]
- Speel, E.J.; Hopman, A.H.; Komminoth, P. Amplification Methods to Increase the Sensitivity of In Situ Hybridization: Play CARD(S). J. Histochem. Cytochem. 1999, 47, 281–288. [Google Scholar] [CrossRef]
- Čmoková, A.; Kolařík, M.; Dobiáš, R.; Hoyer, L.L.; Janouškovcová, H.; Kano, R.; Kuklová, I.; Lysková, P.; Machová, L.; Maier, T.; et al. Resolving the taxonomy of emerging zoonotic pathogens in the Trichophyton benhamiae complex. Fungal Divers. 2020, 104, 333–387. [Google Scholar] [CrossRef]
- Mignon, B.; Swinnen, M.; Bouchara, J.P.; Hofinger, M.; Nikkels, A.; Pierard, G.; Gerday, C.; Losson, B. Purification and characterization of a 315 kDa keratinolytic subtilisin-like serine protease from Microsporum canis and evidence of its secretion in naturally infected cats. Med. Mycol. 1998, 36, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Bitencourt, T.A.; Macedo, C.; Franco, M.E.; Assis, A.F.; Komoto, T.T.; Stehling, E.G.; Beleboni, R.O.; Malavazi, I.; Marins, M.; Fachin, A.L. Transcription profile of Trichophyton rubrum conidia grown on keratin reveals the induction of an adhesin-like protein gene with a tandem repeat pattern. BMC Genom. 2016, 17, 249. [Google Scholar] [CrossRef] [Green Version]
- Bibel, D.J.; A Crumrine, D.; Yee, K.; King, R.D. Development of arthrospores of Trichophyton mentagrophytes. Infect. Immun. 1977, 15, 958–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenhahn, J.; Bartosch, T.; Baumbach, C.-M.; Suchowski, M.; Kacza, J.; Schrödl, W.; Michler, J.K. Detection of subtilisin 3 and 6 in skin biopsies of cattle with clinically manifested bovine ringworm. Med. Mycol. 2021, 59, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Jousson, O.; Léchenne, B.; Bontems, O.; Mignon, B.; Reichard, U.; Barblan, J.; Quadroni, M.; Monod, M. Secreted subtilisin gene family in Trichophyton rubrum. Gene 2004, 339, 79–88. [Google Scholar] [CrossRef]
- Zaugg, C.; Jousson, O.; Léchenne, B.; Staib, P.; Monod, M. Trichophyton rubrum secreted and membrane-associated carboxypeptidases. Int. J. Med. Microbiol. 2008, 298, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Burmester, A.; Shelest, E.; Glöckner, G.; Heddergott, C.; Schindler, S.; Staib, P.; Heidel, A.; Felder, M.; Petzold, A.; Szafranski, K.; et al. Comparative and functional genomics provide insights into the pathogenicity of dermatophytic fungi. Genome Biol. 2011, 12, R7–R16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méhul, B.; Gu, Z.; Jomard, A.; Laffet, G.; Feuilhade, M.; Monod, M. Sub6 (Tri r 2), an Onychomycosis Marker Revealed by Proteomics Analysis of Trichophyton rubrum Secreted Proteins in Patient Nail Samples. J. Investig. Dermatol. 2016, 136, 331–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Niu, Q.; Yu, X.; Jia, X.; Wang, J.; Lin, D.; Jin, Y. Assessment of the function of SUB6 in the pathogenic dermatophyte Trichophyton mentagrophytes. Med. Mycol. 2016, 54, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Woodfolk, J.A.; Wheatley, L.M.; Piyasena, R.V.; Benjamin, D.C.; Platts-Mills, T.A.E. Trichophyton Antigens Associated with IgE Antibodies and Delayed Type Hypersensitivity. Sequence homology to two families of serine proteinases. J. Biol. Chem. 1998, 273, 29489–29496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jousson, O.; Léchenne, B.; Bontems, O.; Capoccia, S.; Mignon, B.; Barblan, J.; Quadroni, M.; Monod, M. Multiplication of an ancestral gene encoding secreted fungalysin preceded species differentiation in the dermatophytes Trichophyton and Microsporum. Microbiology 2004, 150, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Giddey, K.; Monod, M.; Barblan, J.; Potts, A.; Waridel, P.; Zaugg, A.C.; Quadroni, M. Comprehensive Analysis of Proteins Secreted by Trichophyton rubrum and Trichophyton violaceum under in Vitro Conditions. J. Proteome Res. 2007, 6, 3081–3092. [Google Scholar] [CrossRef] [PubMed]
- Giddey, K.; Favre, B.; Quadroni, M.; Monod, M. Closely related dermatophyte species produce different patterns of secreted proteins. FEMS Microbiol. Lett. 2007, 267, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preuett, B.L.; Schuenemann, E.; Brown, J.T.; Kovac, M.E.; Krishnan, S.K.; Abdel-Rahman, S.M. Comparative analysis of secreted enzymes between the anthropophilic–zoophilic sister species Trichophyton tonsurans and Trichophyton equinum. Fungal Biol. 2010, 114, 429–437. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Probe | Target Gene (NCBI acc. no.) | Dye | Sequence (5′-3′) | Tm [°C] | Ref. |
|---|---|---|---|---|---|
| Oligo1 (Tben) | 18S rRNA T. benhamiae (AY083225.1) | 6FAM | CCATGTAGTAAGGTACTATCAA | 60 | own |
| Oligo2 (Tben) | 18S rRNA T. benhamiae (AY083225.1) | 6FAM | TTCGGCAAATCCAAGAATTTCA | 60 | own |
| Oligo3 (Gp) | 18S rRNA Guinea pig (AAKN02059112.1) | Cy3 | TACTACCGATTGGATGGTTTAG | 62 | own |
| Oligo4 (Gp) | 18S rRNA Guinea pig (AAKN02059112.1) | Cy3 | TCTTAGTTGGTGGAGCGATTTG | 64 | own |
| sub3-f | sub3 (AY437854.1) | 6FAM | GAGCAACGCTAACACCCTGGGCAAGCATG | 82 | own |
| sub3-taq | sub3 (AY437854.1) | 6FAM | CAATCTGCTTCAAGCGGTCGCAGGCCT | 86 | own |
| sub6-f | sub6 (AY437857.1) | 6FAM | TACCAGAGAGAGTATCAGTGCTGCCGC | 84 | own |
| sub6-taq | sub6 (AY437857.1) | 6FAM | CCGCAAACGTGAGGAGAAGCCATGGAAG | 88 | own |
| mcpA-f | mcpA (XM_003014418.1) | 6FAM | GGAGTTCCATGCACCGCCTTCAATGC | 82 | own |
| mcpA-taq | mcpA (XM_003014418.1) | 6FAM | GGTAGATGGTGTTGCAGATGGGCCCGG | 88 | own |
| GFP | Green Fluorescent Protein (neg) (MN513050.1) | 6FAM | GAGTTAAAAGGTATTGATTTTAAAG | 88 | own |
| Prev | 16S rRNA Escherichia coli (neg) | FITC | CCACATGTTCCTCCGCTTGT | 62 | [29] |
| PanF-1 | fungi (pos) | 6FAM | CCGATCCCTAGTCGGCATAG | 62 | [19] |
| PanF-2 | fungi (pos) | 6FAM | CTCTGGCTTCACCCTATTC | 58 | [17] |
| EUB 338 | 16S rRNA eubacteria (neg) | 6FAM | GCTGCCTCCCGTAGGAGT | 60 | [28] |
| EUK 516 | 18S rRNA eukaryotes (pos) | 6FAM | ACCAGACTTGCCCTCC | 52 | [28] |
| Gene of Interest [5] | Plasmid (pJET1.2/Blunt; 2974 bp) | Linearity [Molecules/Reaction] | Sensitivity [Molecules/ Reaction] | Efficiency (Validity ± 5%) | Tm Product [°C] |
|---|---|---|---|---|---|
| Isocitrate lyase(isol) | pJET1.2/blunt-Isol (3077 bp) | 50 | 5 | 1.03 | 80.2 |
| Metallocarboxy-peptidase A (mcpA) | pJET1.2/blunt-MCPA (3058 bp) | 500 | 5 | 0.94 | 80.7 |
| Subtilisin 3 (sub 3) | pJET1.2/blunt-Sub3 (3055 bp) | 50 | 5 | 0.99 | 84.0 |
| Subtilisin 6 (sub 6) | pJET1.2/blunt-Sub6 (3095 bp) | 50 | 5 | 1.00 | 82.3 |
| Reference gene: ADP—ribosylation factor (ADPrf) | pJET1.2/blunt-ADPRF (3048 bp) | 50 | 5 | 0.91 | 80.2 |
| Sample Day | T. benhamiae Isolates | |||
|---|---|---|---|---|
| sub 3 | sub 6 | mcpA | isol | |
| 3 (n = 6) | 4.19 × 103 | 1.63 × 103 | 1.65 × 103 | 1.57 × 103 |
| 5 (n = 11) | 4.76 × 103 | 1.14 × 103 | 1.33 × 103 | 1.16 × 103 |
| 7 (n = 13) | 1.25 × 104 | 6.04 × 102 | 1.75 × 103 | 9.03 × 102 |
| 10 (n = 11) | 5.75 × 104 | 8.99 × 102 | 9.17 × 103 | 3.36 × 102 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumbach, C.-M.; Rückner, A.; Partusch, L.; Engel, E.; Schrödl, W.; Michler, J.K. Comprehensive Assessment of the Virulence Factors sub 3, sub 6 and mcpA in the Zoonotic Dermatophyte Trichophyton benhamiae Using FISH and qPCR. J. Fungi 2022, 8, 24. https://doi.org/10.3390/jof8010024
Baumbach C-M, Rückner A, Partusch L, Engel E, Schrödl W, Michler JK. Comprehensive Assessment of the Virulence Factors sub 3, sub 6 and mcpA in the Zoonotic Dermatophyte Trichophyton benhamiae Using FISH and qPCR. Journal of Fungi. 2022; 8(1):24. https://doi.org/10.3390/jof8010024
Chicago/Turabian StyleBaumbach, Christina-Marie, Antje Rückner, Lena Partusch, Eric Engel, Wieland Schrödl, and Jule Kristin Michler. 2022. "Comprehensive Assessment of the Virulence Factors sub 3, sub 6 and mcpA in the Zoonotic Dermatophyte Trichophyton benhamiae Using FISH and qPCR" Journal of Fungi 8, no. 1: 24. https://doi.org/10.3390/jof8010024