
Macrofungi Cultivation in Shady Forest Areas Significantly Increases Microbiome Diversity, Abundance and Functional Capacity in Soil Furrows


Abstract
:1. Introduction
2. Materials and Methods
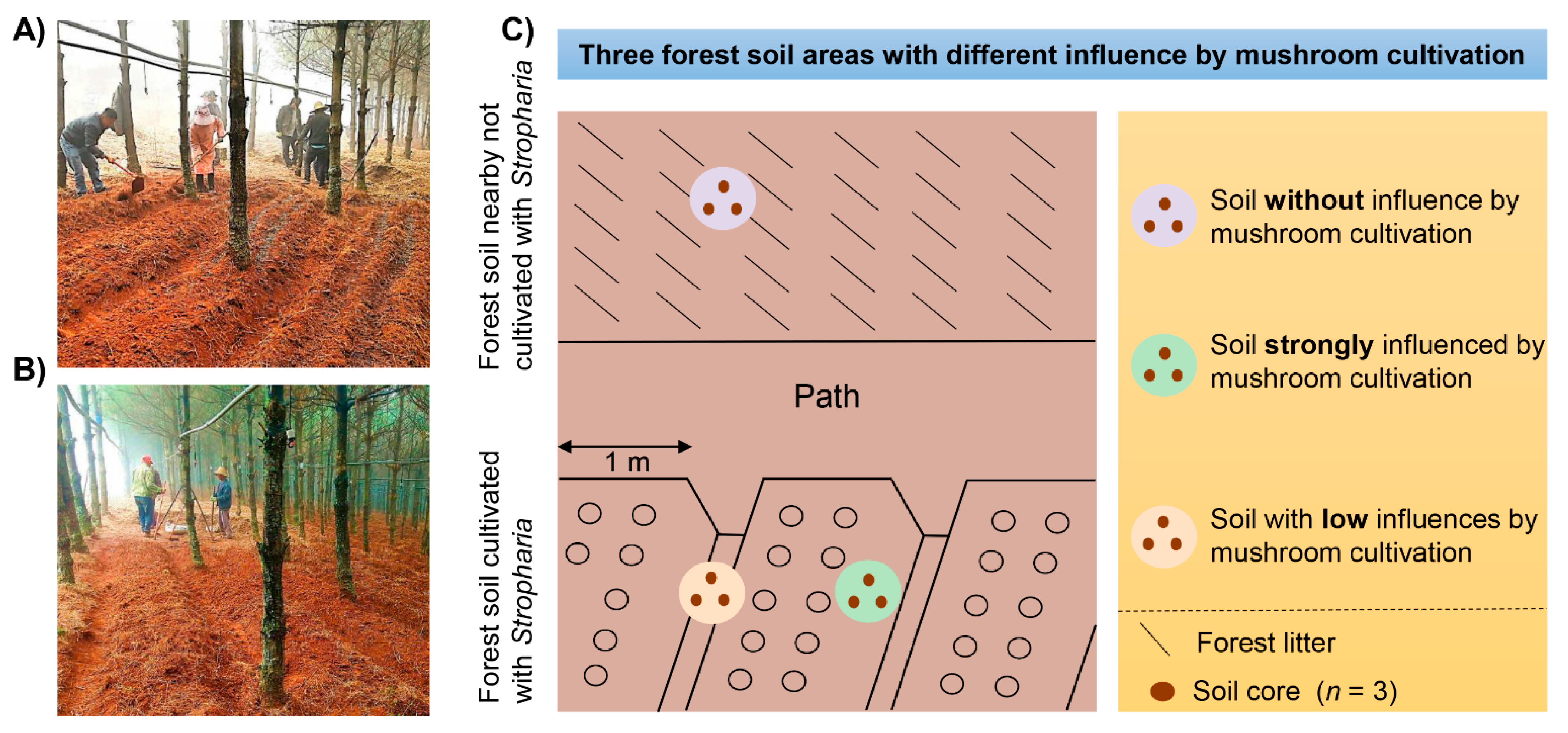
2.1. Site Description and Sampling Method
2.2. Soil Sampling and Chemical Analysis
2.3. DNA Extraction, PCR Amplification and High-Throughput Sequencing
2.4. Quantitative Real-Time PCR
2.5. Statistical Analysis
2.6. Microbial Putative Functional Profile Analysis
3. Results
3.1. Cultivation of Macrofungi Changes Soil Properties
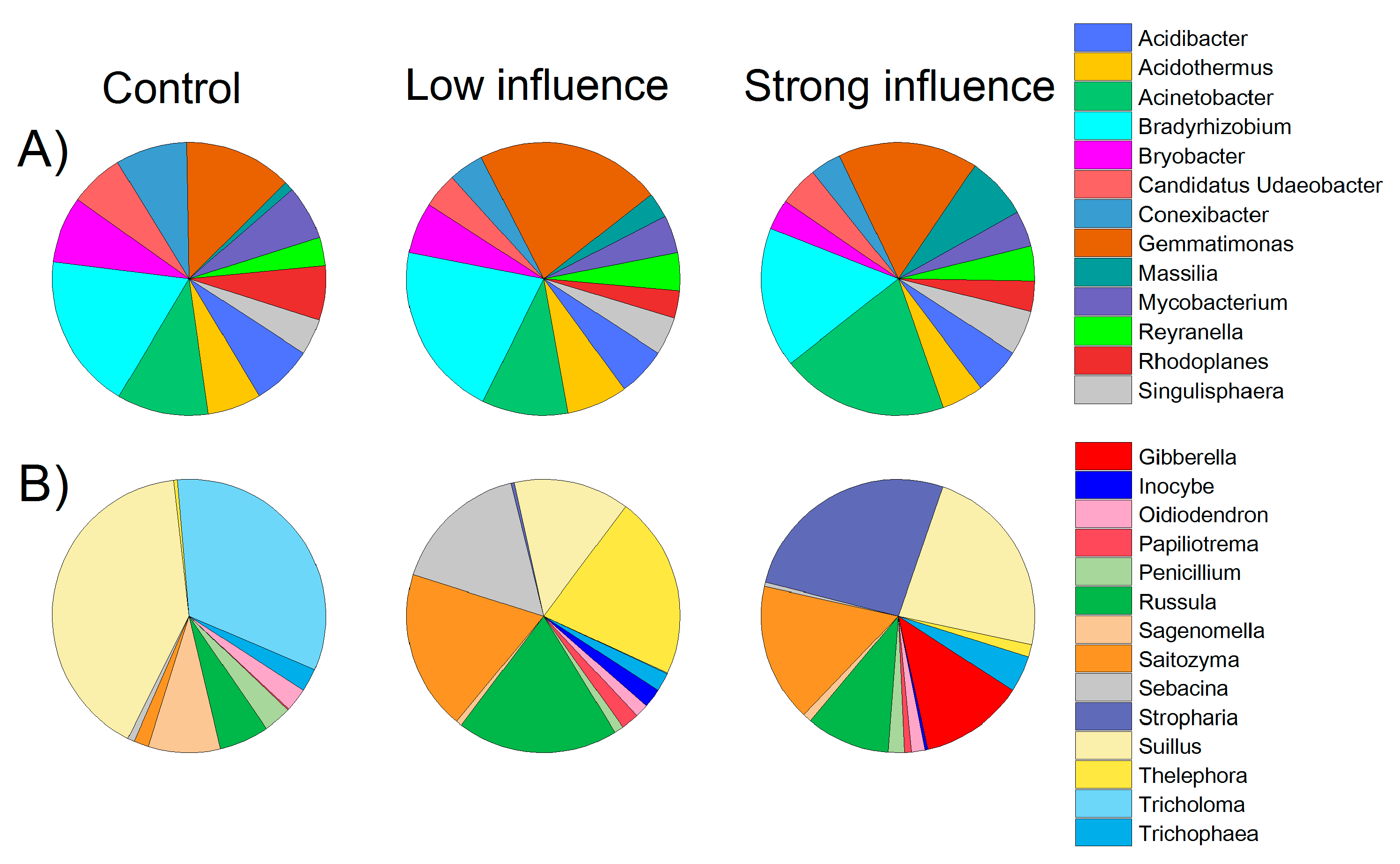
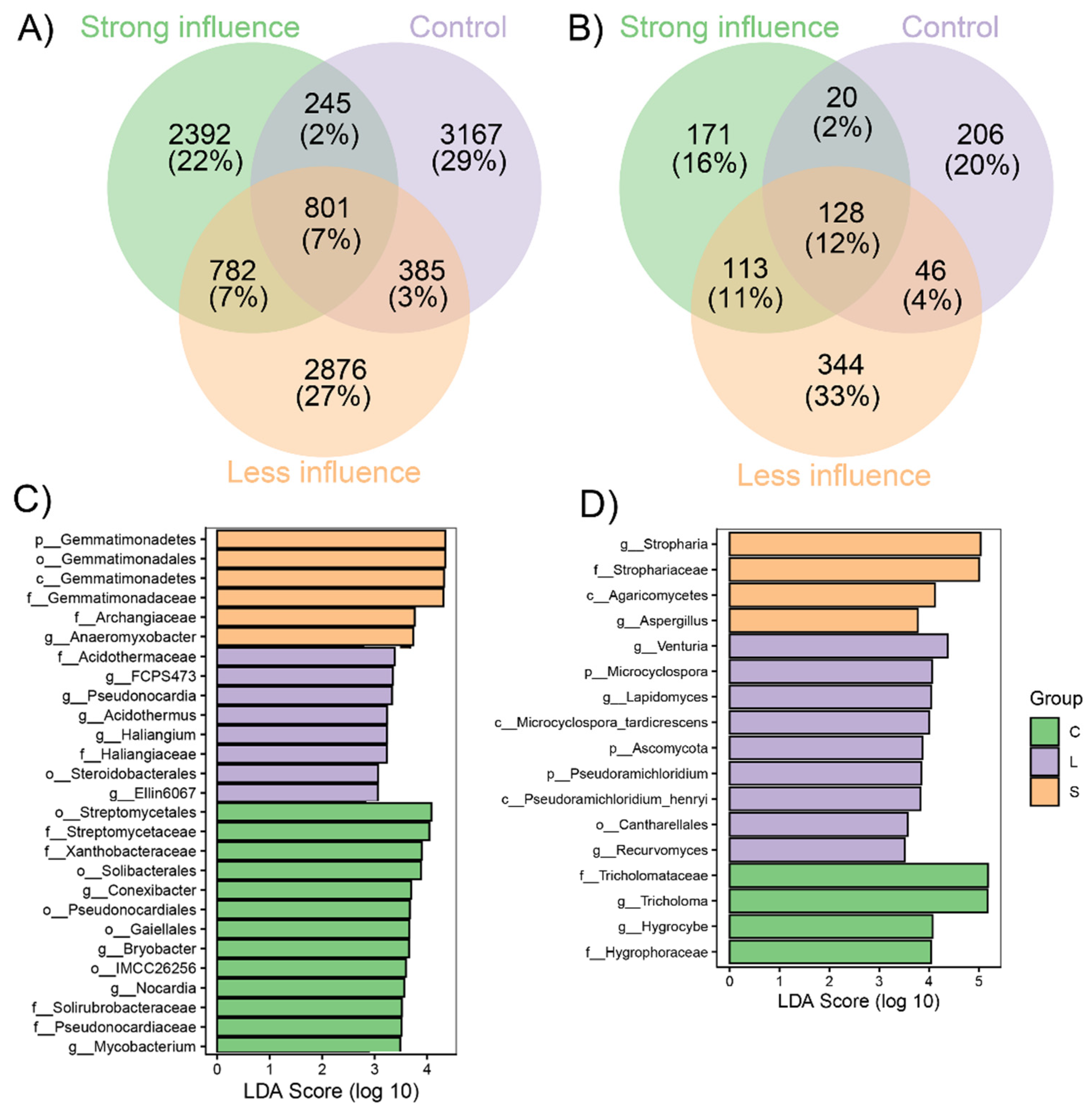
3.2. Various Distribution of Bacterial and Fungal Taxa and Phylotypes
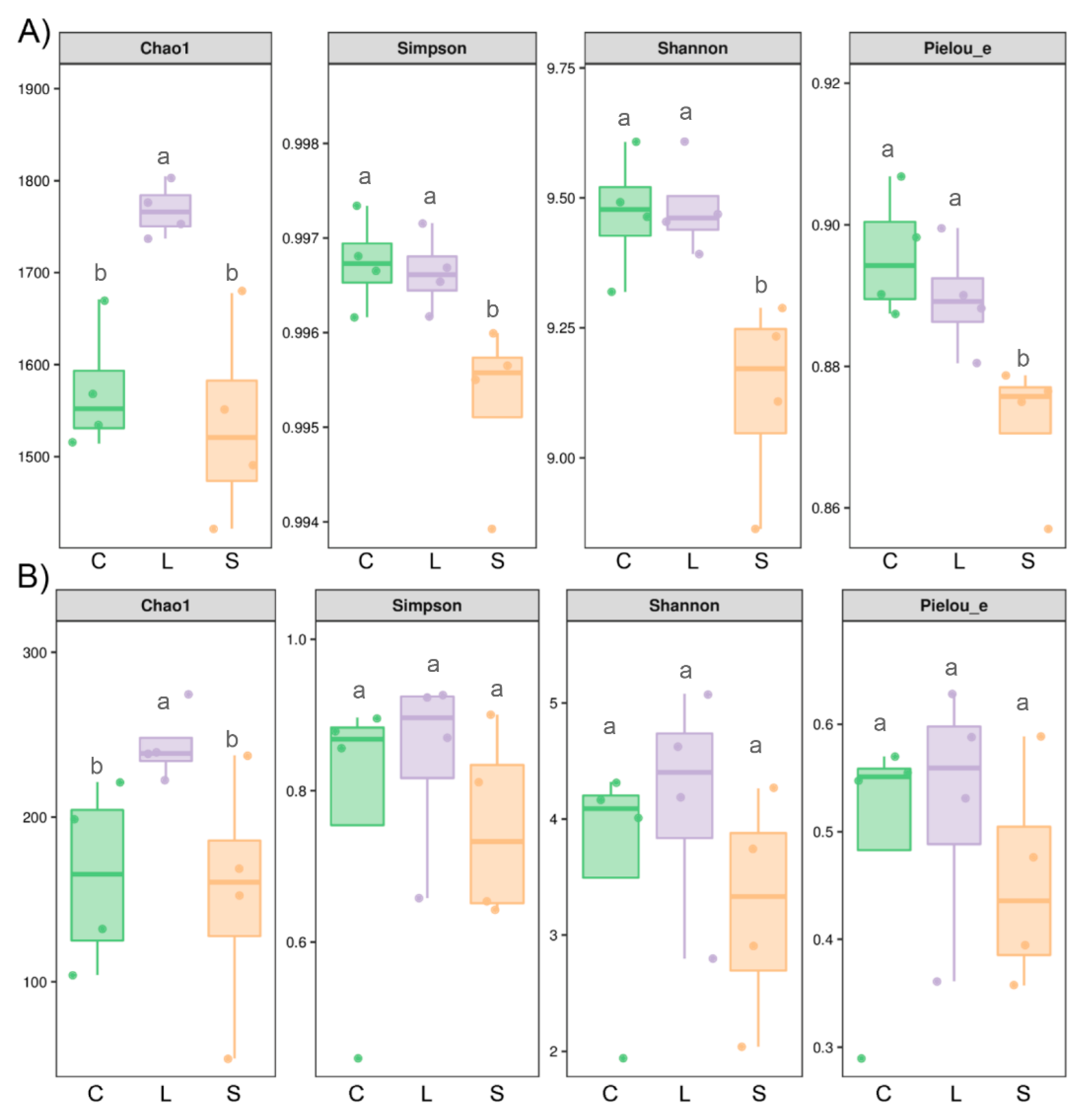
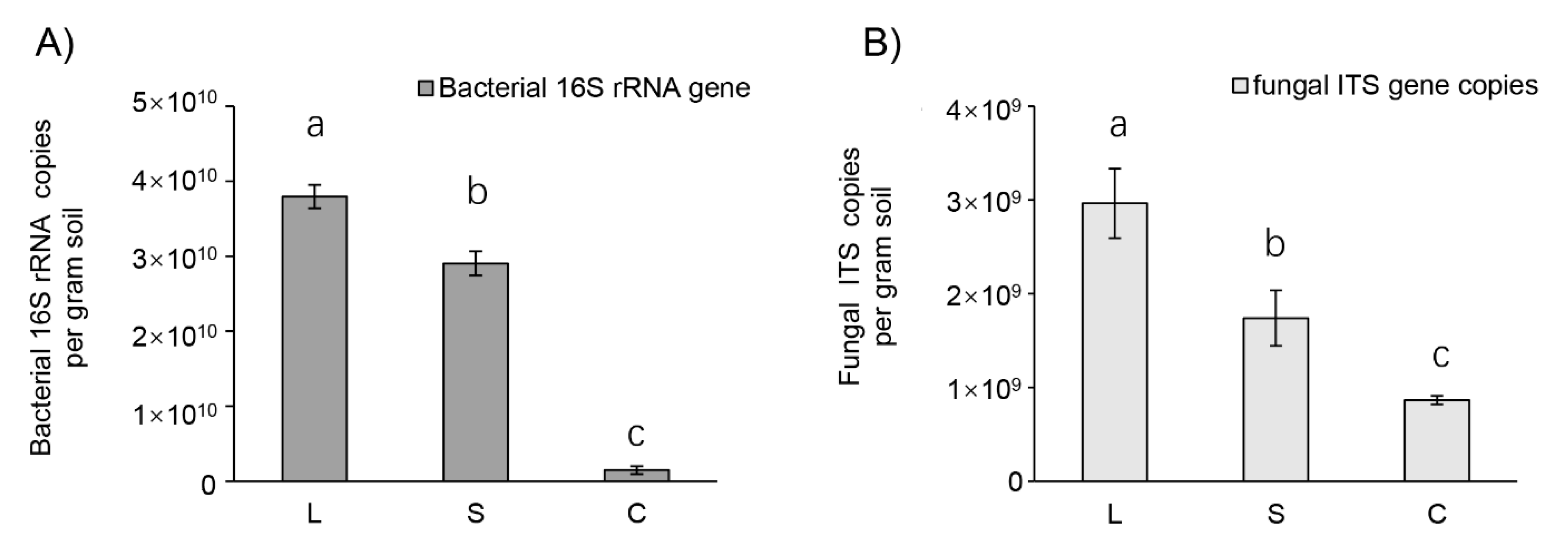
3.3. Bacterial but Not Fungal Diversity Showed Significant Changes
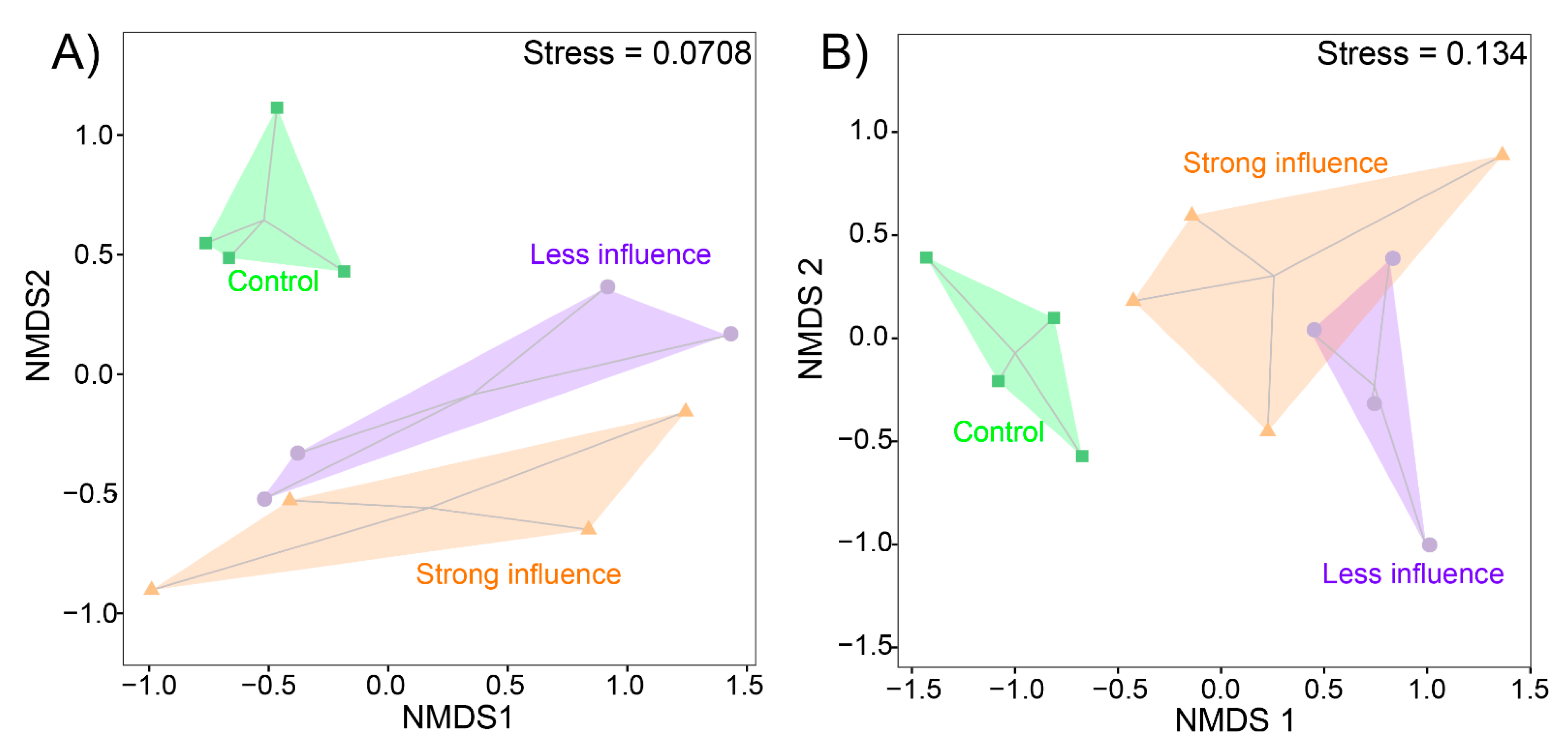
3.4. Both Bacterial and Fungal Community Structure Showed Significant Changes
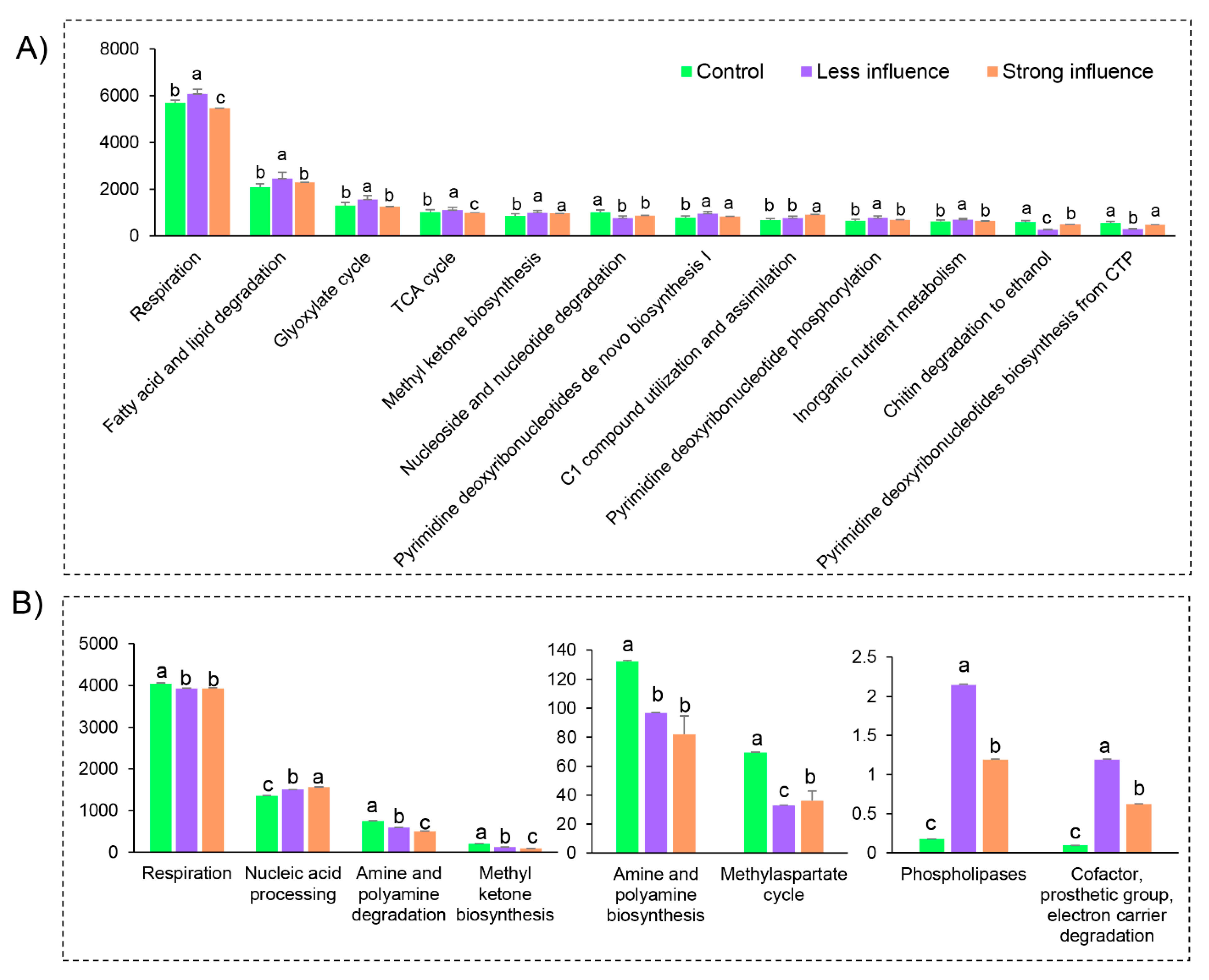
3.5. Microbiome Function Prediction
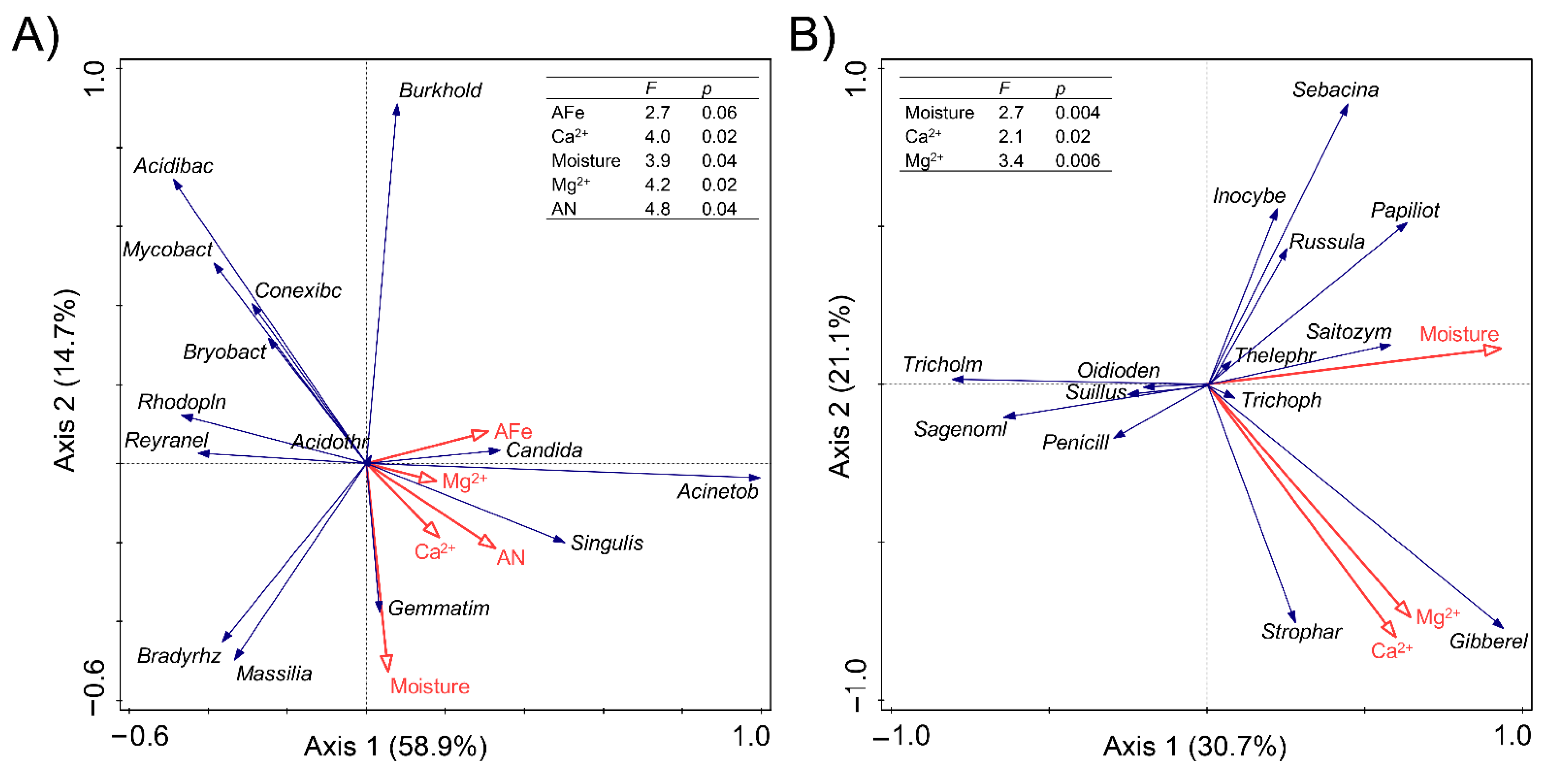
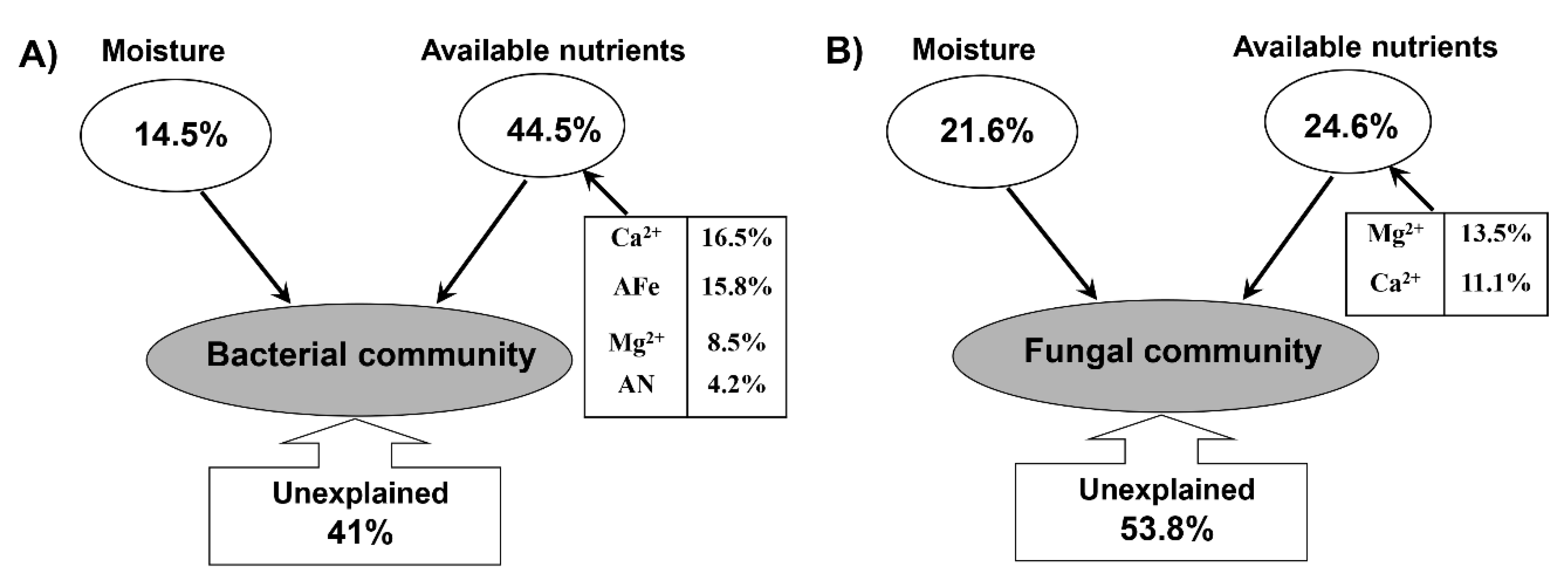
3.6. Correlation between Soil Properties and Microbial Communities
4. Discussion
4.1. Effect of Macrofungi Cultivation on Soil Properties
4.2. Effects of Macrofungi Cultivation on Soil Microbial Communities
4.3. Changes in the Relationship between Microbiome and Soil Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Govorushko, S.; Rezaee, R.; Dumanov, J.; Tsatsakis, A. Poisoning associated with the use of mushrooms: A review of the global pattern and main characteristics. Food Chem. Toxicol. 2019, 128, 267–279. [Google Scholar] [CrossRef]
- Lu, H.; Lou, H.; Hu, J.; Liu, Z.; Chen, Q. Macrofungi: A review of cultivation strategies, bioactivity, and application of mushrooms. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2333–2356. [Google Scholar] [CrossRef]
- Murrill, W.A. Dark-spored agarics: II. Gomphidius and Stropharia. Mycologia 1922, 14, 121. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Kirk, P.M.; Sutton, B.C.; Pegler, D.N. Ainsworth & Bisby’s dictionary of the fungi. Rev. Inst. Med. Trop. Sao Paulo 1996, 38, 272. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Tian, G.; Geng, X.; Zhao, Y.; Ng, T.; Zhao, L.; Wang, H. Isolation and characterization of a novel lectin from the edible mushroom Stropharia rugosoannulata. Molecules 2014, 19, 19880–19891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Yu, S.; Ma, Y.; Huang, H. Integrated economic and environmental analysis of agricultural straw reuse in edible fungi industry. PeerJ 2018, 6, e4624. [Google Scholar] [CrossRef] [Green Version]
- Qing, Y. Cultivation techniques of Stropharia rugosoannulate under forest with high quality, high yield and high efficiency. Mordern Agric. Technol. 2020, 4, 75–77. (In Chinese) [Google Scholar]
- Li, C.L.; Zhang, G.P.; Zhao, Y.J.; Li, H.P. Study on the cultivation of Stropharia rugosoannulate in alpine region of Qinghai province. Agric. Technol. Equip. 2018, 6, 45–46. (In Chinese) [Google Scholar]
- Gong, S.; Chen, C.; Zhu, J.; Qi, G.; Jiang, S. Effects of wine-cap Stropharia cultivation on soil nutrients and bacterial communities in forestlands of northern China. PeerJ 2018, 6, e5741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadiri, M.; Mustapha, Y. The use of spent mushroom substrate of L. subnudus Berk as a soil condition for vegetables. Bayero J. Pure Appl. Sci. 2011, 3, 63212. [Google Scholar] [CrossRef] [Green Version]
- Uzun, I. Use of spent mushroom compost in sustainable fruit production. J. Fruit Ornam. Plant. Res. 2004, 12, 157–165. [Google Scholar]
- Jonathan, S.G.; Lawal, M.M.; Oyetunji, O.J. Effect of spent mushroom compost of Pleurotus pulmonarius on growth performance of four nigerian vegetables. Mycobiology 2011, 39, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, Y.; Lin, X.; Zhang, J.; Zeng, J. Dissipation of polycyclic aromatic hydrocarbons (PAHs) in soil microcosms amended with mushroom cultivation substrate. Soil Biol. Biochem. 2012, 47, 191–197. [Google Scholar] [CrossRef]
- Nazir, R.; Warmink, J.A.; Boersma, H.; van Elsas, J.D. Mechanisms that promote bacterial fitness in fungal-affected soil microhabitats. FEMS Microbiol. Ecol. 2010, 71, 169–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wu, J.; Yang, F.; Chen, Q.; Feng, J.; Li, Q.; Zhang, Q.; Wang, W.; Cheng, X. Linkages between soil organic carbon fractions and carbon-hydrolyzing enzyme activities across riparian zones in the Three Gorges of China. Sci. Rep. 2020, 10, 8433. [Google Scholar] [CrossRef]
- Cusack, D.F.; Silver, W.L.; Torn, M.S.; Burton, S.D.; Firestone, M.K. Changes in microbial community characteristics and soil organic matter with nitrogen additions in two tropical forests. Ecology 2011, 92, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Trillas, M.I.; Segarra, G. Chapter 8 Interactions between nonpathogenic fungi and plants. Adv. Bot. Res. 2009, 51, 321–359. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chater, C.C.C.; Yu, F.; Perez-Moreno, J. Tuber pseudohimalayense ascomata-compartments strongly select their associated bacterial microbiome from nearby pine forest soils independently of their maturation stage. Pedobiologia (Jena) 2021, 87–88, 150743. [Google Scholar] [CrossRef]
- Guo, M. Soil sampling and methods of analysis. J. Environ. Qual. 2009, 38, 375. [Google Scholar] [CrossRef]
- Bao, S.D. Soil Agricultural Chemistry Analysis, 3rd ed.; China Agriculture Press: Beijing, China, 2007. [Google Scholar]
- Sparks, D.L.; Page, A.L.; Helmke, P.A.; Loeppert, R.H. Methods of Soil Analysis Part 3—Chemical Methods; Soil Science Society of America, Inc.: Madison, WI, USA, 1996. [Google Scholar]
- Parkinson, J.A.; Allen, S.E. A wet oxidation procedure suitable for the determination of nitrogen and mineral nutrients in biological material. Commun. Soil Sci. Plant Anal. 1975, 6, 1–11. [Google Scholar] [CrossRef]
- Nu, R.K. Soil Agricultural Chemical Analysis; China Agricultural Science and Technology Press: Beijing, China, 1999. [Google Scholar]
- Joshi, M.; Deshpande, J.D. Polymerse chain reaction: Methods, principles and application. Int. J. Biomed. Res. 2011, 2, 81–97. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Zhu, Y.; Wang, B.; Liu, D.; Bai, H.; Jin, L.; Wang, B.; Ruan, H.; Mao, L.; Jin, F.; et al. Effects of nitrogen addition on rhizospheric soil microbial communities of poplar plantations at different ages. For. Ecol. Manag. 2021, 494, 119328. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Liu, D.; He, X.; Chater, C.C.C.; Perez-Moreno, J.; Yu, F. Microbiome community structure and functional gene partitioning in different micro-niches within a sporocarp-forming fungus. Front. Microbiol. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2012, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Huws, S.A.; Edwards, J.E.; Kim, E.J.; Scollan, N.D. Specificity and sensitivity of eubacterial primers utilized for molecular profiling of bacteria within complex microbial ecosystems. J. Microbiol. Methods 2007, 70, 565–569. [Google Scholar] [CrossRef]
- Fujita, S.I.; Senda, Y.; Nakaguchi, S.; Hashimoto, T. Multiplex PCR using internal transcribed spacer 1 and 2 regions for rapid detection and identification of yeast strains. J. Clin. Microbiol. 2001, 39, 3617–3622. [Google Scholar] [CrossRef] [Green Version]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Liu, D.; Yang, J.; Zhu, Z.; Darboux, F.; Jiao, J.; An, S. Effects of forest floor characteristics on soil labile carbon as varied by topography and vegetation type in the Chinese Loess Plateau. Catena 2021, 196, 104825. [Google Scholar] [CrossRef]
- O’Connor, R.J. Multivariate analysis of ecological communities. Trends Ecol. Evol. 1988, 3, 121. [Google Scholar] [CrossRef]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef]
- Hug, L.A.; Castelle, C.J.; Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Frischkorn, K.R.; Williams, K.H.; Tringe, S.G.; Banfield, J.F. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 2013, 1, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elser, J.J.; Sterner, R.W.; Gorokhova, E.; Fagan, W.F.; Markow, T.A.; Cotner, J.B.; Harrison, J.F.; Hobbie, S.E.; Odell, G.M.; Weider, L.W. Biological stoichiometry from genes to ecosystems. Ecol. Lett. 2000, 3, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.W. Elemental stoichiometry of species in ecosystems. In Linking Species & Ecosystems; Springer US: Boston, MA, USA, 1995; pp. 240–252. [Google Scholar]
- Fujii, K.; Uemura, M.; Hayakawa, C.; Funakawa, S.; Kosaki, T.; Ohta, S. Fluxes of dissolved organic carbon in two tropical forest ecosystems of East Kalimantan, Indonesia. Geoderma 2009, 152, 127–136. [Google Scholar] [CrossRef]
- Qualls, R.G. Biodegradability of fractions of dissolved organic carbon leached from decomposing leaf litter. Environ. Sci. Technol. 2005, 39, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, W.H.; Andrews, J.A. Soil respiration and the global carbon cycle. Biogeochemistry 2000, 48, 7–20. [Google Scholar] [CrossRef]
- Zechmeister-Boltenstern, S.; Keiblinger, K.M.; Mooshammer, M.; Peñuelas, J.; Richter, A.; Sardans, J.; Wanek, W. The application of ecological stoichiometry to plant–microbial–soil organic matter transformations. Ecol. Monogr. 2015, 85, 133–155. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, C.C.; Liptzin, D. C:N:P stoichiometry in soil: Is there a “Redfield ratio” for the microbial biomass? Biogeochemistry 2007, 85, 235–252. [Google Scholar] [CrossRef]
- Lindahl, B.D.; Tunlid, A. Ectomycorrhizal fungi—potential organic matter decomposers, yet not saprotrophs. New Phytol. 2015, 205, 1443–1447. [Google Scholar] [CrossRef]
- Comandini, O.; Rinaldi, A.C.; Kuyper, T.W. Measuring and Estimating Ectomycorrhizal Fungal Diversity: A Continuous Challenge; Nova Science Publishers: New York, NY, USA, 2012; pp. 165–200. [Google Scholar]
- Błońska, E.; Lasota, J. β-Glucosidase activity of forest soil as an indicator of soil carbon accumulation. In Soil Biological Communities and Ecosystem Resilience; Springer International Publishing: Cham, Switzerland, 2017; pp. 253–263. [Google Scholar]
- Song, W.; Kim, M.; Tripathi, B.M.; Kim, H.; Adams, J.M. Predictable communities of soil bacteria in relation to nutrient concentration and successional stage in a laboratory culture experiment. Environ. Microbiol. 2016, 18, 1740–1753. [Google Scholar] [CrossRef] [Green Version]
- Baldrian, P. Forest microbiome: Diversity, complexity and dynamics. FEMS Microbiol. Rev. 2017, 41, 109–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z. Influence of calcium cyanamide application on the pathogens and traits of lei bamboo forest soil. Hans J. Soil Sci. 2013, 1, 9–13. [Google Scholar] [CrossRef]
- Hosseini, S.A.; Réthoré, E.; Pluchon, S.; Ali, N.; Billiot, B.; Yvin, J.C. Calcium application enhances drought stress tolerance in sugar beet and promotes plant biomass and beetroot sucrose concentration. Int. J. Mol. Sci. 2019, 20, 3777. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Xiong, J.; Zhang, H.; Feng, Y.; Lin, X.; Li, X.; Liang, W.; Chu, H. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 2013, 57, 204–211. [Google Scholar] [CrossRef]
- Brockett, B.F.T.; Prescott, C.E.; Grayston, S.J. Soil moisture is the major factor influencing microbial community structure and enzyme activities across seven biogeoclimatic zones in western Canada. Soil Biol. Biochem. 2012, 44, 9–20. [Google Scholar] [CrossRef]
- Deepika, S.; Kothamasi, D. Soil moisture—a regulator of arbuscular mycorrhizal fungal community assembly and symbiotic phosphorus uptake. Mycorrhiza 2015, 25, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Erlandson, S.R.; Savage, J.A.; Cavender-Bares, J.M.; Peay, K.G. Soil moisture and chemistry influence diversity of ectomycorrhizal fungal communities associating with willow along an hydrologic gradient. FEMS Microbiol. Ecol. 2016, 92, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierer, N.; Schimel, J.P.; Holden, P.A. Influence of drying-rewetting frequency on soil bacterial community structure. Microb. Ecol. 2003, 45, 63–71. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Moisture | pH | OM | TC | TN | TP | C:N | C:P | N:P |
|---|---|---|---|---|---|---|---|---|---|
| (%) | (g kg−1) | (g kg−1) | (g kg−1) | (g kg−1) | |||||
| S | 29.4(1.7) a | 5.0(0.04) b | 389(36) a | 226(21) a | 4.6(0.2) a | 1.06(0.05) a | 48.9(4.2) a | 214(24) a | 4.4(0.2) a |
| L | 31.4(2.0) a | 5.1(0.06) ab | 76.5(11) b | 44.4(6.7) b | 2.4(0.4) b | 0.95(0.10) a | 18.8(1.9) b | 46.9(5.2) b | 2.5(0.4) b |
| C | 20.8(0.2) b | 5.2(0.01) a | 74.4(15) b | 43.2(8.7) b | 2.5(0.4) b | 0.88(0.09) ab | 17.5(2.0) b | 48.7(6.5) b | 2.8(0.2) b |
| Treatment | Ca2+ | Mg2+ | AN | AP | AMn | AFe |
|---|---|---|---|---|---|---|
| (mg kg−1) | (mg kg−1) | (mg kg−1) | (mg kg−1) | (mg kg−1) | (mg kg−1) | |
| S | 2934(399) a | 1437(341) a | 369(28) a | 25(4.1) a | 211(53) a | 216(19) a |
| L | 1017(122) b | 80(32) b | 232(33) b | 6.5(1.7) b | 29.3(15) b | 38.1(5.9) b |
| C | 973(121) b | 74(27) b | 225(44) b | 5.2(0.8) c | 24.3(12) b | 33.1(7.4) b |
| Treatment | ACP | Glucosidase | Cellobiosidase | Peroxidase |
|---|---|---|---|---|
| (nmol g−1 h−1) | (nmol g−1 h−1) | (nmol g−1 h−1) | (nmol g−1 h−1) | |
| S | 1005(246) a | 94.4(7.1) a | 20.6(2.3) a | 1613(12) a |
| L | 746(480) b | 77.5(6.8) b | 14.1(2.0) b | 1557(77) a |
| C | 603(85) c | 64.0(6.6) b | 13.7(1.5) b | 1510(121) a |
| Group | Bacterial Community | Fungal Community | |||
|---|---|---|---|---|---|
| Pseudo-F b | p-Value | Pseudo-F | p-Value | ||
| Overall a | 1.509 | 0.032 | 2.903 | 0.002 | |
| PERMANOVA | C vs. L c | 1.743 | 0.036 | 4.821 | 0.031 |
| C vs. S | 2.017 | 0.036 | 2.962 | 0.041 | |
| L vs. S | 0.842 | 0.546 | 1.469 | 0.140 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Wang, Y.; Zhang, P.; Yu, F.; Perez-Moreno, J. Macrofungi Cultivation in Shady Forest Areas Significantly Increases Microbiome Diversity, Abundance and Functional Capacity in Soil Furrows. J. Fungi 2021, 7, 775. https://doi.org/10.3390/jof7090775
Liu D, Wang Y, Zhang P, Yu F, Perez-Moreno J. Macrofungi Cultivation in Shady Forest Areas Significantly Increases Microbiome Diversity, Abundance and Functional Capacity in Soil Furrows. Journal of Fungi. 2021; 7(9):775. https://doi.org/10.3390/jof7090775
Chicago/Turabian StyleLiu, Dong, Yanliang Wang, Peng Zhang, Fuqiang Yu, and Jesús Perez-Moreno. 2021. "Macrofungi Cultivation in Shady Forest Areas Significantly Increases Microbiome Diversity, Abundance and Functional Capacity in Soil Furrows" Journal of Fungi 7, no. 9: 775. https://doi.org/10.3390/jof7090775