
Using High-Throughput Amplicon Sequencing to Evaluate Intragenomic Variation and Accuracy in Species Identification of Cordyceps Species


Abstract
:1. Introduction
2. Materials and Methods
2.1. The Sample Collection and DNA Extraction
2.2. PacBio Amplicon Sequencing
2.3. Sanger Sequencing
2.4. Phylogenetic Analyses
2.5. Sequence Divergence Analysis
2.6. Haplotype Network Analysis of nrITS
3. Results
3.1. PacBio Sequencing
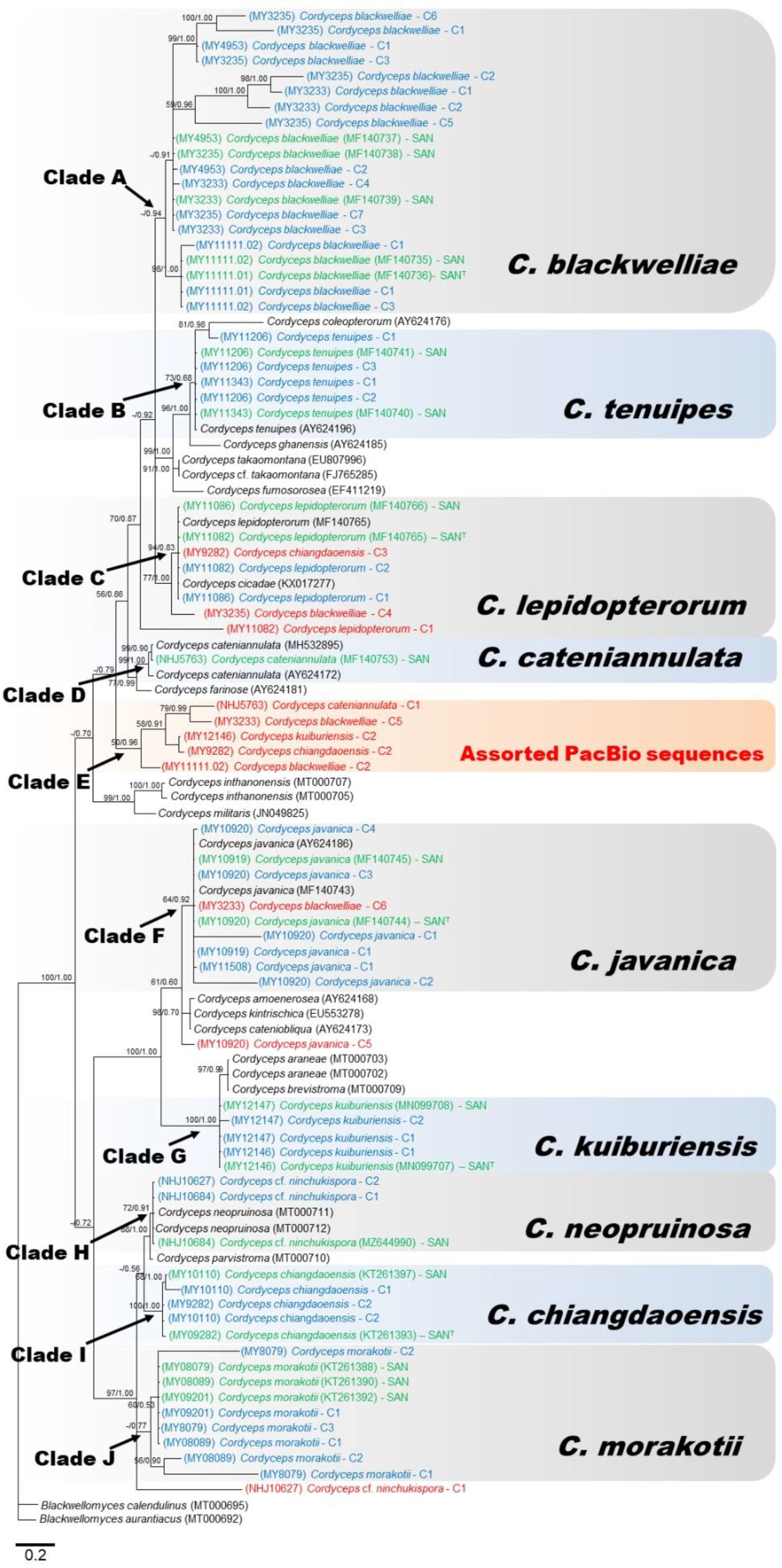
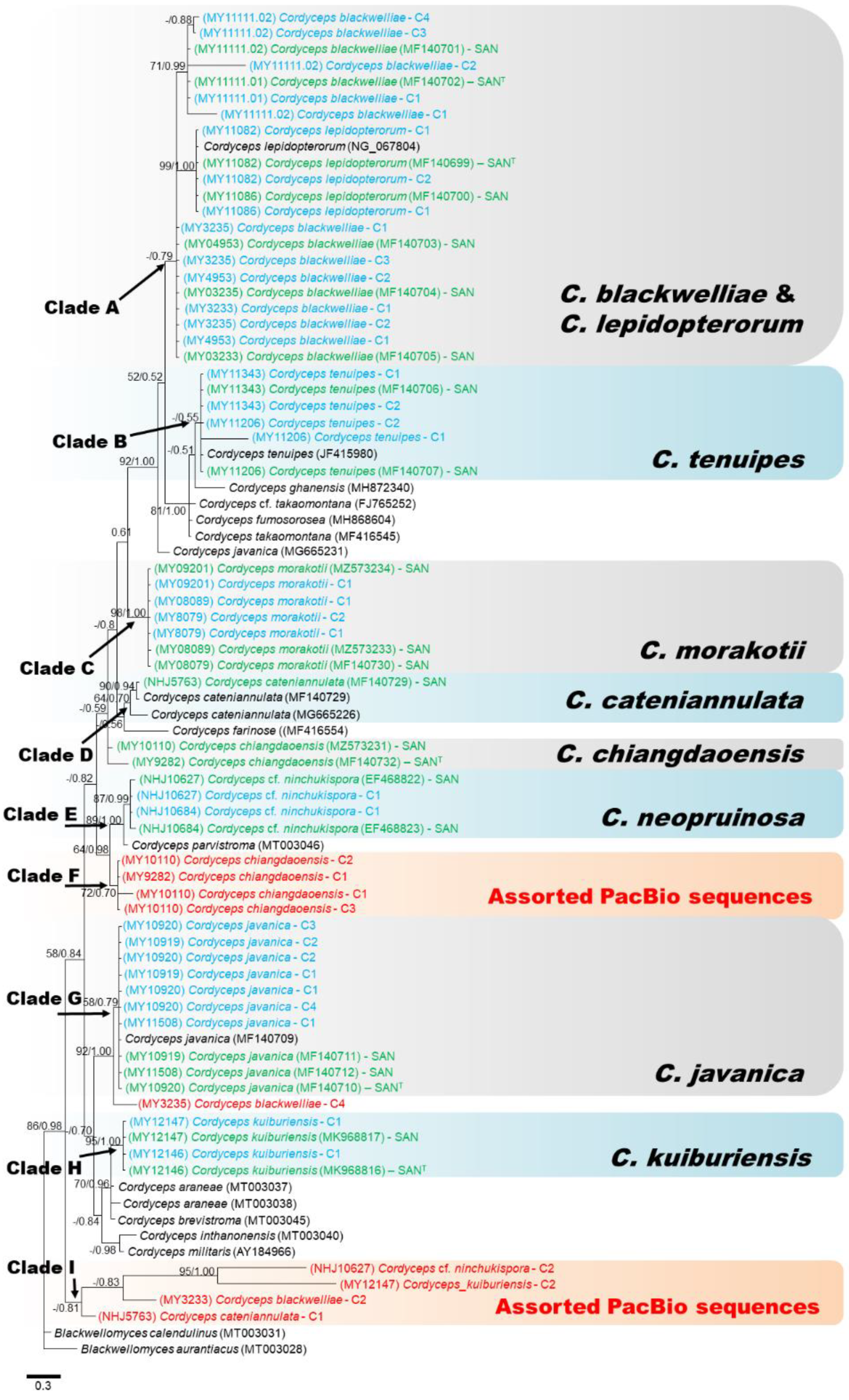
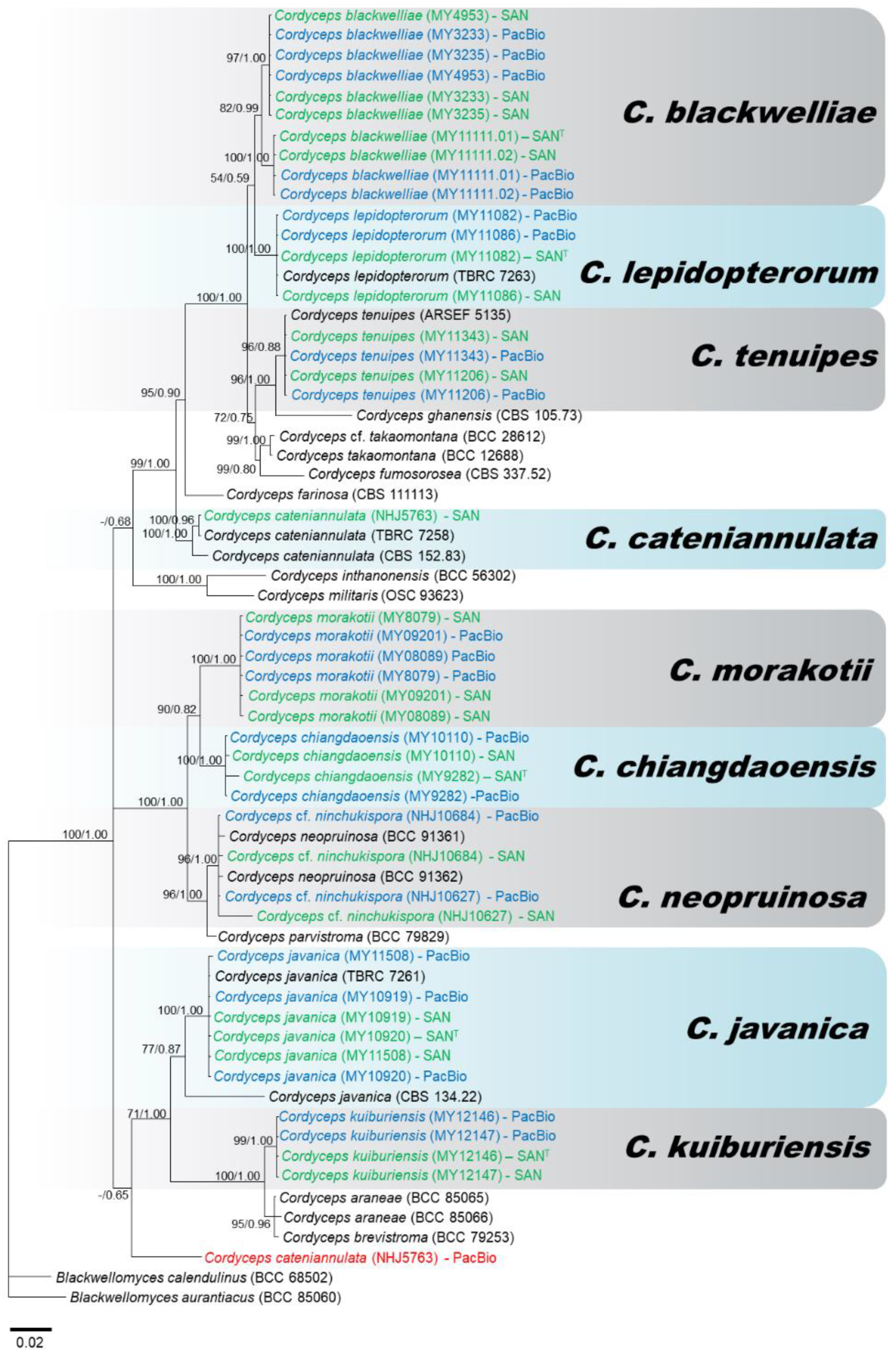
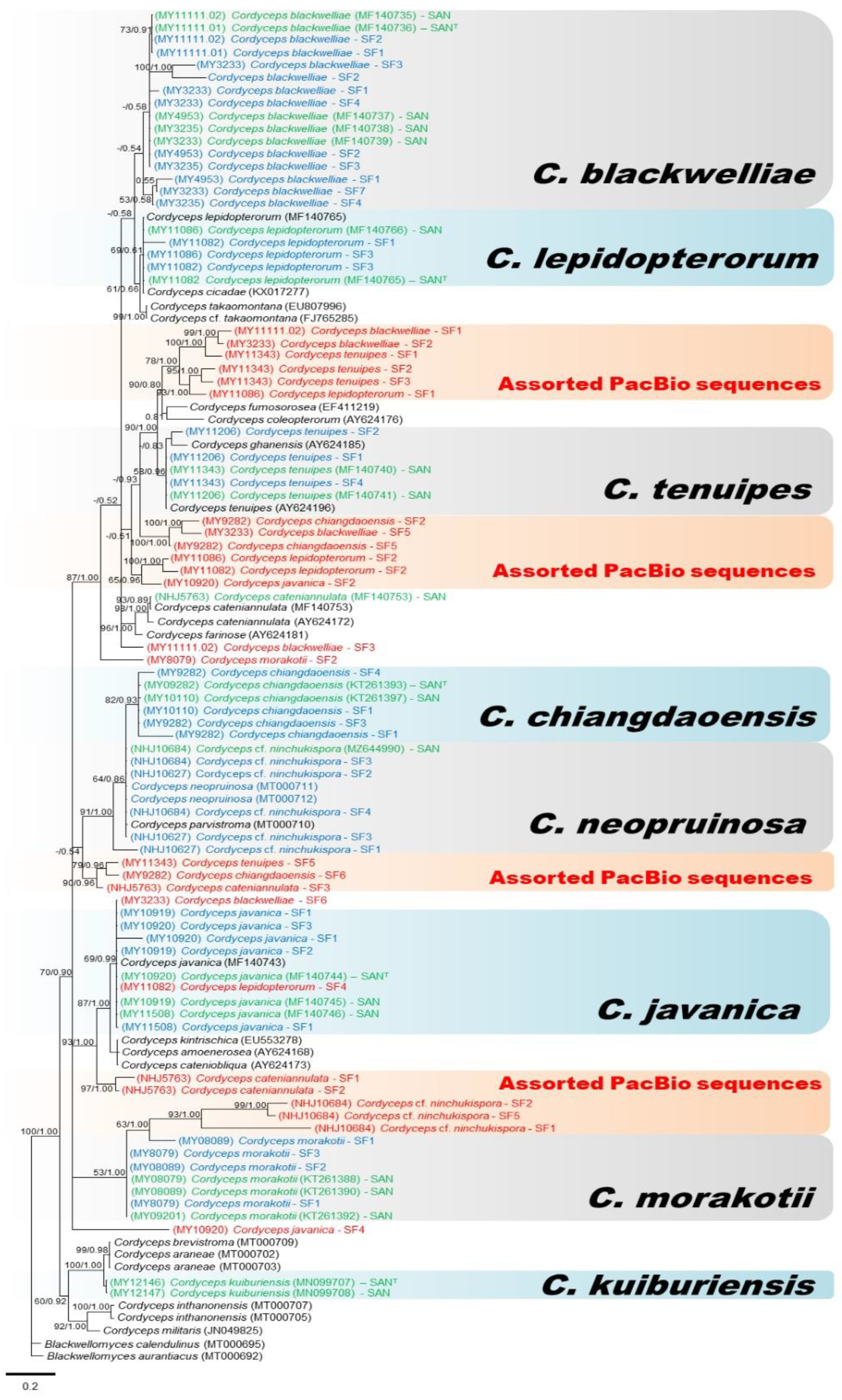
3.2. Molecular Phylogeny
3.3. Sequence Divergence Analysis
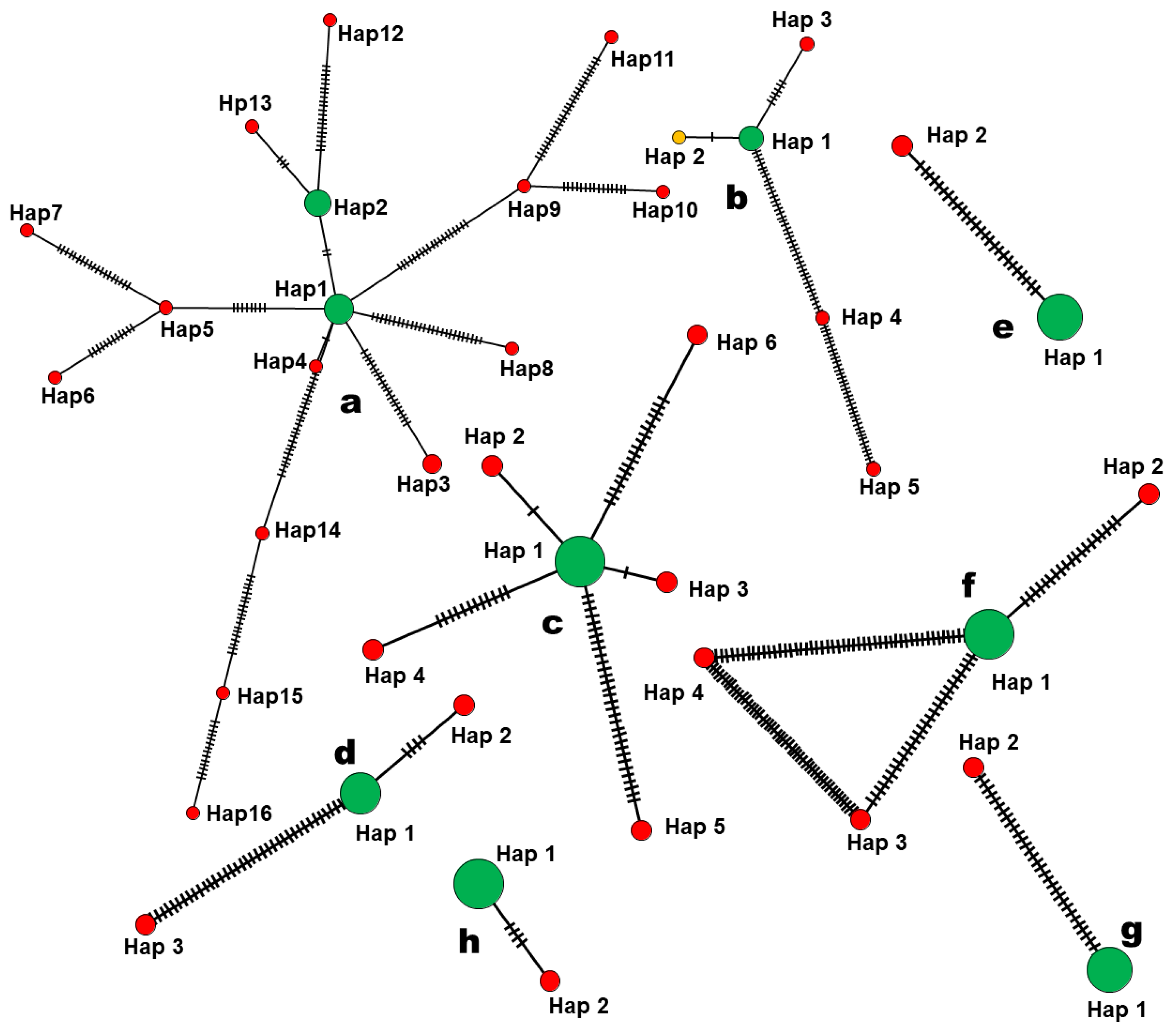
3.4. Haplotype Network Analysis of nrITS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, M.; De Cal, A.; Melgarejo, P.; Larena, I.; Espeso, E.A. The development of genetic and molecular markers to register and commercialize Penicillium rubens (formerly Penicillium oxalicum) strain 212 as a biocontrol agent. Microb. Biotechnol. 2016, 9, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Badotti, F.; de Oliveira, F.S.; Garcia, C.F.; Vaz, A.B.M.; Fonseca, L.C.; Nahum, L.A.; Oliveira, G.; Góes-Neto, A. Effectiveness of ITS and sub-regions as DNA barcode markers for the identification of Basidiomycota (Fungi). BMC Microbiol. 2017, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Mongkolsamrit, S.; Khonsanit, A.; Thanakitpipattana, D.; Tasanathai, K.; Noisripoom, W.; Lamlertthon, S.; Himaman, W.; Houbraken, J.; Samson, R.A.; Luangsa-ard, J. Revisiting Metarhizium and the description of new species from Thailand. Stud. Mycol. 2020, 95, 171–251. [Google Scholar] [CrossRef]
- Baturo-Cieśniewska, A.; Pusz, W.; Patejuk, K. Problems, Limitations, and Challenges in Species Identification of Ascomycota Members on the Basis of ITS Regions. Acta Mycol. 2020, 55, 5512. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.H. Intraspecific ITS variability in the Kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 2008, 4, 193–201. [Google Scholar] [CrossRef]
- Arnold, A.E.; Miadlikowska, J.; Higgins, K.L.; Sarvate, S.D.; Gugger, P.; Way, A.; Hofstetter, V.; Kauff, F.; Lutzoni, F. A phylogenetic estimation of trophic transition networks for ascomycetous fungi: Are lichens cradles of symbiotrophic fungal diversification? Syst. Biol. 2009, 58, 283–297. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, M. The fungi: 1, 2, 3 … 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef]
- Liu, K.-L.; Porras-Alfaro, A.; Kuske, C.R.; Eichorst, S.A.; Xie, G. Accurate, rapid taxonomic classification of fungal large-subunit rRNA genes. Appl. Environ. Microbiol. 2012, 78, 1523–1533. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Shukla, P.K. Use of PCR targeting of internal transcribed spacer regions and single-stranded conformation polymorphism analysis of sequence variation in different regions of rRNA genes in fungi for rapid diagnosis of mycotic keratitis. J. Clin. Microbial. 2005, 43, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Begerow, D.; Nilsson, H.; Unterseher, M.; Maier, W. Current state and perspectives of fungal DNA barcoding and rapid identification procedures. Appl. Microbiol. Biotechnol. 2010, 87, 99–108. [Google Scholar] [CrossRef]
- Porras-Alfaro, A.; Liu, K.L.; Kuske, C.R.; Xie, G. From Genus to Phylum: Large-Subunit and Internal Transcribed Spacer rRNA Operon Regions Show Similar Classification Accuracies Influenced by Database Composition. Appl. Environ. Microbiol. 2014, 80, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilgalys, R.; Hester, M. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 1990, 172, 4238–4246. [Google Scholar] [CrossRef] [Green Version]
- Bruns, T.D.; Shefferson, R.P. Evolutionary studies of ectomycorrhizal fungi: Milestones and future directions. Can. J. Bot. 2004, 82, 1122–1132. [Google Scholar] [CrossRef]
- Nei, M.; Rooney, A.P. Concerted and Birth-and-Death Evolution of Multigene Families. Annu. Rev. Genet. 2005, 39, 121–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, K.W.; Petersen, R.H. Apparent Recombination or Gene Conversion in the Ribosomal ITS Region of a Flammulina (Fungi, Agaricales) Hybrid. Mol. Biol. Evol. 2001, 18, 94–96. [Google Scholar] [CrossRef] [Green Version]
- Hughes, K.W.; Petersen, R.H.; Lodge, D.J.; Bergemann, S.E.; Baumgartner, K.; Tulloss, R.E.; Lickey, E.; Cifuentes, J. Evolutionary consequences of putative intra-and interspecific hybridization in agaric fungi. Mycologia 2013, 105, 1577–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vydryakova, G.A.; Van, D.T.; Shoukouhi, P.; Psurtseva, N.V.; Bissett, J. Intergenomic and intragenomic ITS sequence heterogeneity in Neonothopanus nambi (Agaricales) from Vietnam. Mycology 2012, 3, 89–99. [Google Scholar] [CrossRef]
- Batovska, J.; Cogan, N.O.; Lynch, S.E.; Blacket, M.J. Using Next-Generation Sequencing for DNA Barcoding: Capturing Allelic Variation in ITS2. G3 Genes Genomes Genet. 2017, 7, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef]
- Index Fungorum. Available online: www.indexfungorum.org (accessed on 20 April 2021).
- Sung, G.H.; Hywel-Jones, N.L.; Sung, J.M.; Luangsa-Ard, J.J.; Shrestha, B.; Spatafora, J.W. Phylogenetic classification of Cordyceps and the clavicipitaceous fungi. Stud. Mycol. 2007, 57, 5–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongkolsamrit, S.; Noisripoom, W.; Tasanathai, K.; Khonsanit, A.; Thanakitpipattana, D.; Himaman, W.; Kobmoo, N.; Luangsa-ard, J.J. Molecular phylogeny and morphology reveal cryptic species in Blackwellomyces and Cordyceps (Cordycipitaceae) from Thailand. Mycol. Prog. 2020, 19, 957–983. [Google Scholar] [CrossRef]
- Tasanathai, K.; Thanakitpipattana, D.; Noisripoom, W.; Khonsanit, A.; Kumsao, J.; Luangsa-ard, J.J. Two new Cordyceps species from a community forest in Thailand. Mycol. Prog. 2016, 15, 28. [Google Scholar] [CrossRef]
- Mongkolsamrit, S.; Noisripoom, W.; Thanakitpipattana, D.; Wutikhun, T.; Spatafora, J.W.; Luangsa-ard, J.J. Disentangling cryptic species with isaria-like morphs in Cordycipitaceae. Mycologia 2018, 110, 230–257. [Google Scholar] [CrossRef]
- Crous, P.W.; Wingfield, M.J.; Lombard, L.; Roets, F.; Swart, W.J.; Alvarado, P.; Carnegie, A.J.; Moreno, G.; Luangsa-ard, J.J.; Thangavel, R.; et al. Fungal planet description sheets: 951–1041. Persoonia 2019, 43, 223–425. [Google Scholar] [CrossRef]
- Tedersoo, L.; Drenkhan, R.; Anslan, S.; Morales-Rodriguez, C.; Cleary, M. High-throughput identification and diagnostics of pathogens and pests: Overview and practical recommendations. Mol. Ecol. Resour. 2019, 19, 47–76. [Google Scholar] [CrossRef] [Green Version]
- Mongkolsamrit, S.; Luangsa-ard, J.J.; Spatafora, J.W.; Sung, G.H.; Hywel-Jones, N.L. A combined ITS rDNA and beta-tubulin phylogeny of Thai species of Hypocrella with nonfragmenting ascospores. Mycol. Res. 2009, 113, 684–699. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: London, UK, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- GitHub. Available online: https://github.com/PacificBiosciences/ccs (accessed on 6 September 2020).
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paloi, S.; Mhuanthong, W.; Kobmoo, N. “PacBio amplicon sequencing of Cordyceps species”, Mendeley Data, V2. 2021. [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X Windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Holder, M.T.; Vos, R.; Midford, P.E.; Liebowitz, T.; Chan, L.; Hoover, P.; Warnow, T. The CIPRES Portals. 2010. Available online: http://www.phylo.org/sub_sections/portal (accessed on 17 March 2021).
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.W.; Bryant, D. PopArt: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Paradis, E. Analysis of haplotype networks: The randomized minimum spanning tree method. Methods Ecol. Evol. 2018, 9, 1308–1317. [Google Scholar] [CrossRef] [Green Version]
- Estensmo, E.L.F.; Maurice, S.; Morgado, L.; Martin-Sanchez, P.M.; Skrede, I.; Kauserud, H. The influence of intraspecific sequence variation during DNA metabarcoding: A case study of eleven fungal species. Mol. Ecol. Resour. 2021, 21, 1141–1148. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Braukmann, T.W.A.; Prosser, S.W.J.; Ratnasingham, S.; deWaard, J.R.; Ivanova, N.V.; Janzen, D.H.; Hallwachs, W.; Naik, S.; Sones, J.E.; et al. A Sequel to Sanger: Amplicon sequencing that scales. BMC Genom. 2018, 19, 219. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Tooming-Klunderud, A.; Anslan, S. PacBio metabarcoding of Fungi and other eukaryotes: Errors, biases and perspectives. New Phytol. 2018, 217, 1370–1385. [Google Scholar] [CrossRef] [Green Version]
- Wurzbacher, C.; Larsson, E.; Bengtsson-Palme, J.; den Wyngaert, S.V.; Svantesson, S.; Kristiansson, E.; Kagami, M.; Nilsson, R.H. Introducing ribosomal tandem repeat barcoding for fungi. Mol. Ecol. Resour. 2019, 19, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Wang, A.; Au, K.F. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 2019, 20, 26. [Google Scholar] [CrossRef] [Green Version]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, T.J.; Hunter, R.; et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, 34, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Colabella, C.; Corte, L.; Roscini, L.; Bassetti, M.; Tascini, C.; Mellor, J.C.; Meyer, W.; Robert, V.; Vu, D.; Cardinali, G. NGS barcode sequencing in taxonomy and diagnostics, an application in “Candida” pathogenic yeasts with a metagenomic perspective. IMA Fungus 2018, 9, 91–105. [Google Scholar] [CrossRef]
- Filges, S.; Yamada, E.; Ståhlberg, A.; Godfrey, T.E. Impact of Polymerase Fidelity on Background Error Rates in Next-Generation Sequencing with Unique Molecular Identifiers/Barcodes. Sci. Rep. 2019, 9, 3503. [Google Scholar] [CrossRef] [PubMed]
- Hestand, M.S.; Van Houdt, J.; Cristofoli, F.; Vermeesch, J.R. Polymerase specific error rates and profiles identified by single molecule sequencing. Mutat. Res. 2016, 784, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Potapov, V.; Ong, J.L. Correction: Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE 2017, 12, e0181128. [Google Scholar] [CrossRef] [Green Version]
- Kårén, O.; Högberg, N.; Dahlberg, A.; Jonsson, L.; Nylund, J. Inter- and intraspecific variation in the ITS region of rDNA of ectomycorrhizal fungi in Fennoscandia detected by endonuclease analysis. New Phytol. 1997, 136, 313–325. [Google Scholar] [CrossRef]
- Horton, T.R. Molecular approaches to ectomycorrhizal diversity studies: Variation in ITS at a local scale. Plant Soil 2002, 244, 29–39. [Google Scholar] [CrossRef]
- Lindner, D.L.; Banik, M.T. Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in genus Laetiporus. Mycologia 2011, 103, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Kijpornyongpan, T.; Aime, C.M. Rare or rarely detected? Ceraceosorus guamensis sp. nov.: A second described species of Ceraceosorales and the potential for under detection of rare lineages with common sampling techniques. Antonie Van Leeuwenhoek 2016, 109, 1127–1139. [Google Scholar] [CrossRef]
- Hughes, K.W.; Tulloss, R.H.; Petersen, R.H. Intragenomic nuclear RNA variation in a cryptic Amanita taxon. Mycologia 2018, 110, 93–103. [Google Scholar] [CrossRef]
- Paloi, S.; Luangsa-ard, J.J.; Mhuanthong, W.; Stadler, M.; Kobmoo, N. Intragenomic variation in nuclear ribosomal markers and its implication in species delimitation, identification and barcoding in Fungi. Fungal Biol. Rev. 2021, in press. [Google Scholar]
- Pannecoucque, J.; Höfte, M. Detection of rDNA ITS polymorphism in Rhizoctonia solani AG 2-1 isolates. Mycologia 2009, 101, 26–33. [Google Scholar] [CrossRef]
- Roscini, L.; Tristezza, M.; Corte, L.; Colabella, C.; Perrotta, C.; Rampino, P.; Robert, V.; Vu, D.; Cardinali, G.; Grieco, F. Early ongoing speciation of Ogataea uvarum sp. nov. within the grape ecosystem revealed by the internal variability among the rDNA operon repeats. Front. Microbiol. 2018, 9, 1687. [Google Scholar] [CrossRef]
- Moricca, S.; Kasuga, T.; Mitchelson, K.; Ragazzi, A.; Diamandis, S. Heterogeneity in intergenic regions of the ribosomal repeat of the pine-blister rusts Cronartium flaccidum and Peridermium pini. Curr. Genet. 1996, 29, 388–394. [Google Scholar] [CrossRef]
- Smith, M.E.; Douhan, G.W.; Rizzo, D.M. Intraspecific and intrasporocarp ITS variation of ectomycorrhizal fungi as assessed by rDNA sequencing of sporocarps and pooled ectomycorrhizal roots from Quercus woodland. Mycorrhiza 2007, 18, 15–22. [Google Scholar] [CrossRef]
- Rooney, A.P.; Ward, T.J. Evolution of a large ribosomal RNA multigene family in filamentous fungi: Birth and death of a concerted evolution paradigm. Proc. Natl. Acad. Sci. USA 2005, 102, 5084–5089. [Google Scholar] [CrossRef] [Green Version]
- Simon, U.K.; Weiß, M. Intragenomic variation of fungal ribosomal genes is higher than previously thought. Mol. Biol. Evol. 2008, 25, 2251–2254. [Google Scholar] [CrossRef] [Green Version]
- James, S.A.; O’Kelly, M.J.T.; Carter, D.M.; Davey, R.P.; Van Oudenaarden, A.; Roberts, I.N. Repetitive sequence variation and dynamics in the ribosomal DNA array of Saccharomyces cerevisiae as revealed by whole-genome resequencing. Genome Res. 2009, 19, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.; Lambert, C.; Wibberg, D.; Kalinowski, J.; Cox, R.J.; Kolařík, M.; Kuhnert, E. Intragenomic polymorphisms in the ITS region of high-quality genomes of the Hypoxylaceae (Xylariales, Ascomycota). Mycol. Prog. 2020, 19, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Original Code | BCC Code | TBRC Code | BBH Code | ITS Accession | LSU Accession |
|---|---|---|---|---|---|---|
| C. blackwelliae | MY3233 | BCC 30924 | TBRC 7253 | BBH 23883 | MF140739 | MF140705 |
| MY3235 | BCC 30926 | TBRC 7254 | BBH 23885 | MF140738 | MF140704 | |
| MY4953 | BCC 37652 | TBRC 7255 | BBH 26339 | MF140737 | MF140703 | |
| MY11111.01 T | BCC 79714 | TBRC 7256 | BBH 40750 | MF140736 | MF140702 | |
| MY11111.02 | BCC 79855 | TBRC 7257 | BBH 40750 | MF140735 | MF140701 | |
| C. cateniannulata | NHJ5763 | BCC 1856 | TBRC 7258 | - | MF140753 | MF140729 |
| C. chiangdaoensis | MY9282 T | BCC 68469 | TBRC 7274 | - | KT261393 | MF140732 |
| MY10110 | BCC 75733 | - | - | KT261397 | MZ573231 * | |
| C. javanica | MY10919 | BCC 79245 | TBRC 7259 | BBH 40411 | MF140745 | MF140711 |
| MY10920 T | BCC 79246 | TBRC 7260 | BBH 40412 | MF140744 | MF140710 | |
| MY11508 | BCC 82944 | TBRC 7262 | BBH 41986 | MF140746 | MF140712 | |
| C. kuiburiensis | MY12146 T | BCC 90322 | - | BBH 45453 | MN099707 | MK968816 |
| MY12147 | BCC 90323 | - | BBH 45454 | MN099708 | MK968817 | |
| C. lepidopterorum | MY11082 T | BCC 79840 | TBRC 7263 | BBH 40735 | MF140765 | MF140699 |
| MY11086 | BCC 79842 | TBRC 7264 | BBH 40737 | MF140766 | MF140700 | |
| C. morakotii | MY8079 | BCC 55820 | TBRC 7275 | - | KT261388 | MF140730 |
| MY08089 | BCC 55830 | - | - | KT261390 | MZ573233 * | |
| MY09201 | BCC 68403 | - | - | KT261392 | MZ573234 * | |
| Cordyceps cf. ninchukispora | NHJ10627 | BCC 02744 | - | - | - | EF468822 |
| NHJ10684 | BCC 02725 | - | - | MZ644990 * | EF468823 | |
| C. tenuipes | MY11343 | BCC 82079 | TBRC 7267 | BBH 42147 | MF140740 | MF140706 |
| MY11206 | BCC 81469 | TBRC 7265 | BBH 41247 | MF140741 | MF140707 |
| Species | Strains | Intragenomic Variation (PacBio) | Sanger-PacBio Discrepancy | Intraspecific Variation (Sanger) | Intraspecific Variation (PacBio) |
|---|---|---|---|---|---|
| C. blackwelliae | MY3233 | 0.077 | 0.05 | 0.003 | 0.054 |
| MY3235 | 0.083 | 0.056 | |||
| MY4953 | 0.002 | 0.001 | |||
| MY11111.01 | - | 0 | |||
| MY11111.02 | 0.053 | 0.025 | |||
| C. cateniannulata | NHJ5763 | - | 0.078 | - | - |
| C. chiangdaoensis | MY9282 | 0.075 | 0.063 | 0.004 | 0.061 |
| MY10110 | 0.012 | 0.006 | |||
| C. javanica | MY10919 | - | 0 | 0 | 0.034 |
| MY10920 | 0.056 | 0.028 | |||
| MY11508 | - | 0.002 | |||
| C. kuiburiensis | MY12146 | 0.065 | 0.033 | 0.002 | 0.038 |
| MY12147 | 0.007 | 0.003 | |||
| C. lepidopterorum | MY11082 | 0.045 | 0.24 | 0 | 0.03 |
| MY11086 | - | 0 | |||
| C. morakotii | MY8079 | 0.088 | 0.038 | 0 | 0.054 |
| MY08089 | 0.019 | 0.01 | |||
| MY09201 | - | 0 | |||
| C. cf. ninchukispora | NHJ10627 | 0 | - | NA | 0.037 |
| NHJ10684 | - | 0 | |||
| C. tenuipes | MY11343 | - | 0 | 0 | 0.004 |
| MY11206 | 0.009 | 0.004 |
| Species | Haplotype | Frequency | Sequences |
|---|---|---|---|
| C. blackwelliae | Hap 1 | 5 | MY3233 (SAN); MY3235 (SAN); MY4953 (SAN); MY3235-C7 (341); MY4953-C1 (341) |
| Hap 2 | 4 | MY11111.01 (SAN); MY11111.02 (SAN); MY11111.01-C1 (99); MY11111.02-C3 (251) | |
| Hap 3 | 2 | MY4953-C2 (2); MY3233-C3 (328) | |
| Hap 4 | 1 | MY3233-C4 (1) | |
| Hap 5 | 1 | MY3235-C3 (14) | |
| Hap 6 | 1 | MY3235-C6 (1) | |
| Hap 7 | 1 | MY3235-C1 (1) | |
| Hap 8 | 1 | MY3235-C5 (1) | |
| Hap 9 | 1 | MY3235-C4 (1) | |
| Hap 10 | 1 | MY3233-C6 (1) | |
| Hap 11 | 1 | MY3233-C5 (1) | |
| Hap 12 | 1 | MY1111102-C2 (1) | |
| Hap 13 | 1 | MY11111.02-C1 (1) | |
| Hap 14 | 1 | MY3233-C2 (1) | |
| Hap 15 | 1 | MY3233-C1 (1) | |
| Hap 16 | 1 | MY3235-C2 (1) | |
| C. chiangdaoensis | Hap 1 | 3 | MY10110 (SAN); MY9282-C2 (246); MY10110-C2 (207) |
| Hap 2 | 1 | MY09282 (SAN) | |
| Hap 3 | 1 | MY10110-C1 (1) | |
| Hap 4 | 1 | MY9282-C1 (2) | |
| Hap 5 | 1 | MY9282-C3 (1) | |
| C. javanica | Hap 1 | 6 | AY624186 (SAN); MY10919 (SAN); MY10920 (SAN); MF140743 (SAN); MY10919-C1 (241); MY10920-C3 (259) |
| Hap 2 | 1 | MY10920-C4 (1) | |
| Hap 3 | 1 | MY11508-C1 (129) | |
| Hap 4 | 1 | MY10920-C5 (1) | |
| Hap 5 | 1 | MY10920-C2 (1) | |
| Hap 6 | 1 | MY10920-C1 (1) | |
| C. kuiburiensis | Hap 1 | 4 | MY12146 (SAN); MY12147 (SAN); MY12146-C1 (210); MY12147-C1 (215) |
| Hap 2 | 1 | MY12147-C2 (2) | |
| Hap 3 | 1 | MY12146-C2 (1) | |
| C. lepidopterorum | Hap 1 | 5 | MY11082 (SAN); MY11086 (SAN); MF140765 (SAN); MY11082-C2 (295); MY11086-C1 (225) |
| Hap 2 | 1 | MY11082-C1 (2) | |
| C. morakotii | Hap 1 | 6 | MY08089 (SAN); MY08079 (SAN); MY09201 (SAN); MY8079-C3 (265); MY08089-C1 (350); MY09201-C1 (211) |
| Hap 2 | 1 | MY08089-C2 (1) | |
| Hap 3 | 1 | MY8079-C2 (1) | |
| Hap 4 | 1 | MY8079-C1 (1) | |
| C. cf. ninchukispora | Hap 1 | 5 | MT000711 (SAN); MT000712 (SAN); NHJ10684 (SAN); NHJ10627-C2 (95); NHJ10684-C1(186) |
| Hap 2 | 1 | NHJ10627-C1 (1) | |
| C. tenuipes | Hap 1 | 6 | AY624196 (SAN); MY11343 (SAN); MY11206 (SAN); MY11343-C1 (115); MY11206-C2 (124); MY11206-C3 (303) |
| Hap 2 | 1 | MY11206-C1 (1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paloi, S.; Mhuantong, W.; Luangsa-ard, J.J.; Kobmoo, N. Using High-Throughput Amplicon Sequencing to Evaluate Intragenomic Variation and Accuracy in Species Identification of Cordyceps Species. J. Fungi 2021, 7, 767. https://doi.org/10.3390/jof7090767
Paloi S, Mhuantong W, Luangsa-ard JJ, Kobmoo N. Using High-Throughput Amplicon Sequencing to Evaluate Intragenomic Variation and Accuracy in Species Identification of Cordyceps Species. Journal of Fungi. 2021; 7(9):767. https://doi.org/10.3390/jof7090767
Chicago/Turabian StylePaloi, Soumitra, Wuttichai Mhuantong, Janet Jennifer Luangsa-ard, and Noppol Kobmoo. 2021. "Using High-Throughput Amplicon Sequencing to Evaluate Intragenomic Variation and Accuracy in Species Identification of Cordyceps Species" Journal of Fungi 7, no. 9: 767. https://doi.org/10.3390/jof7090767