
Transcription Factors in the Fungus Aspergillus nidulans: Markers of Genetic Innovation, Network Rewiring and Conflict between Genomics and Transcriptomics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Database of the Potential TFs Encoded by the A. nidulans Genome
2.2. Bioinformatic Analyses
2.3. RNA-Seq Data Analyses
2.4. Generation of Recombinant Strains of A. nidulans
3. Results
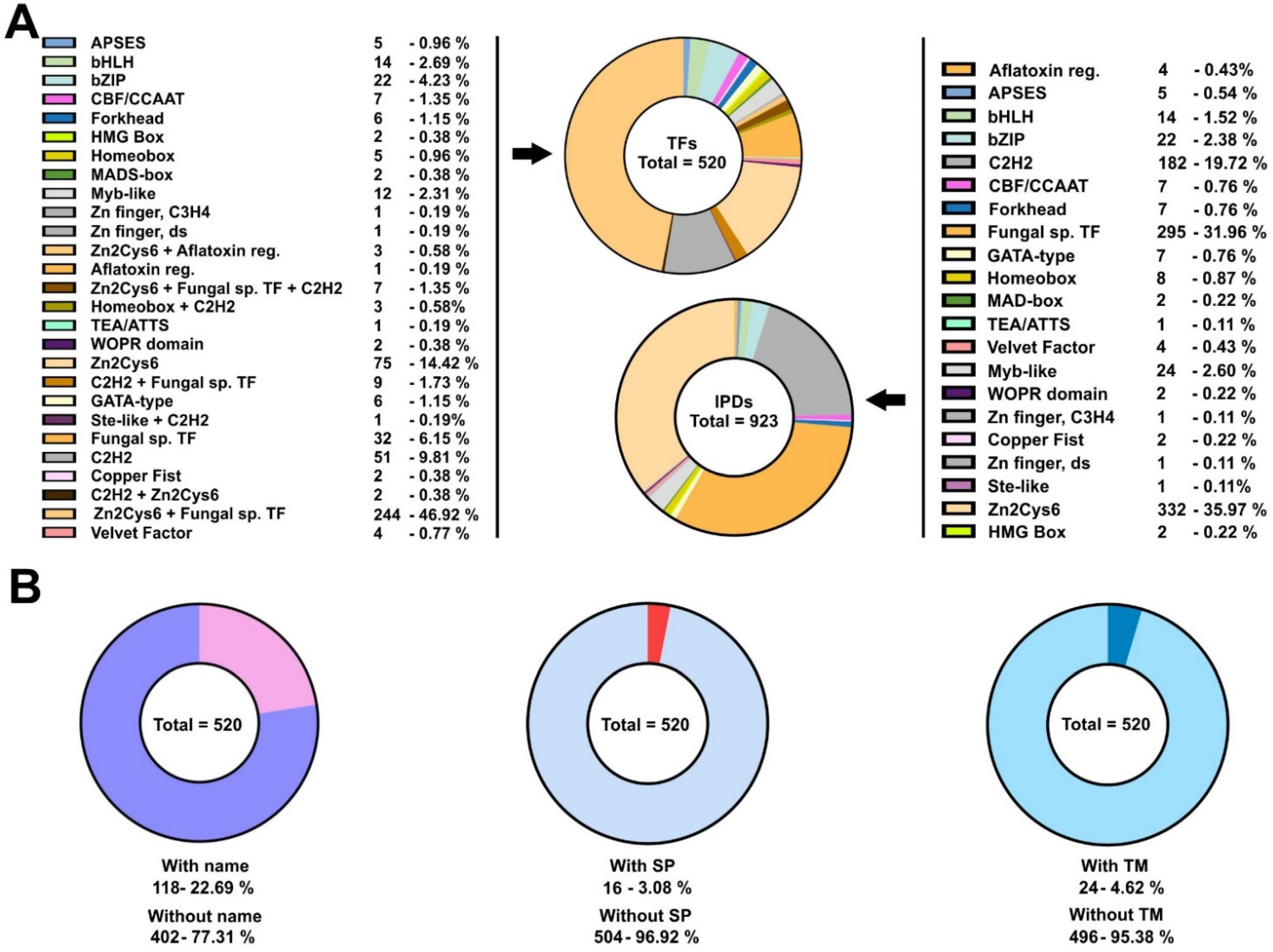
3.1. The Set of Potential TFs in the Ascomycete Fungus Aspergillus nidulans Is Dominated by Binuclear Zinc Clusters
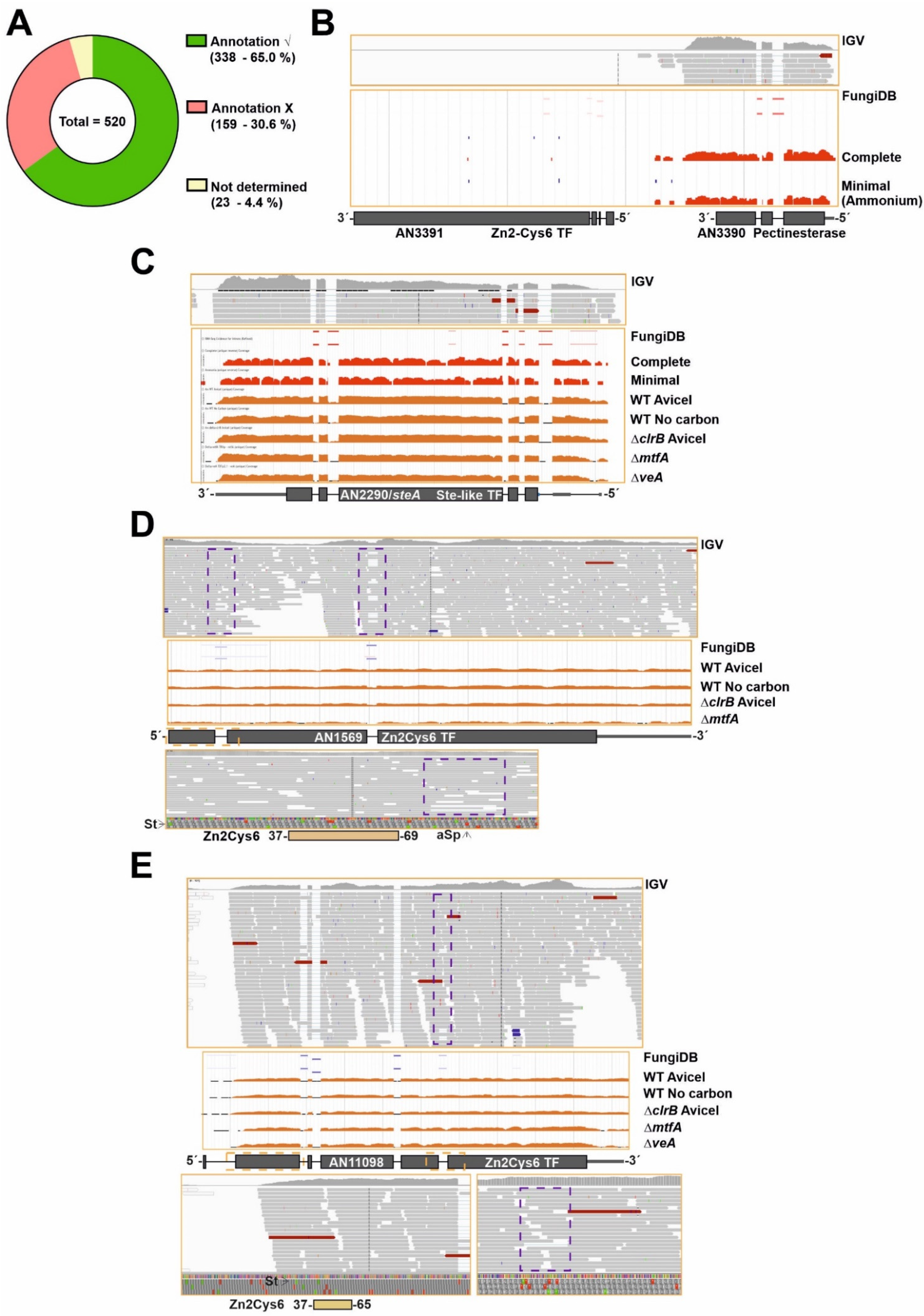
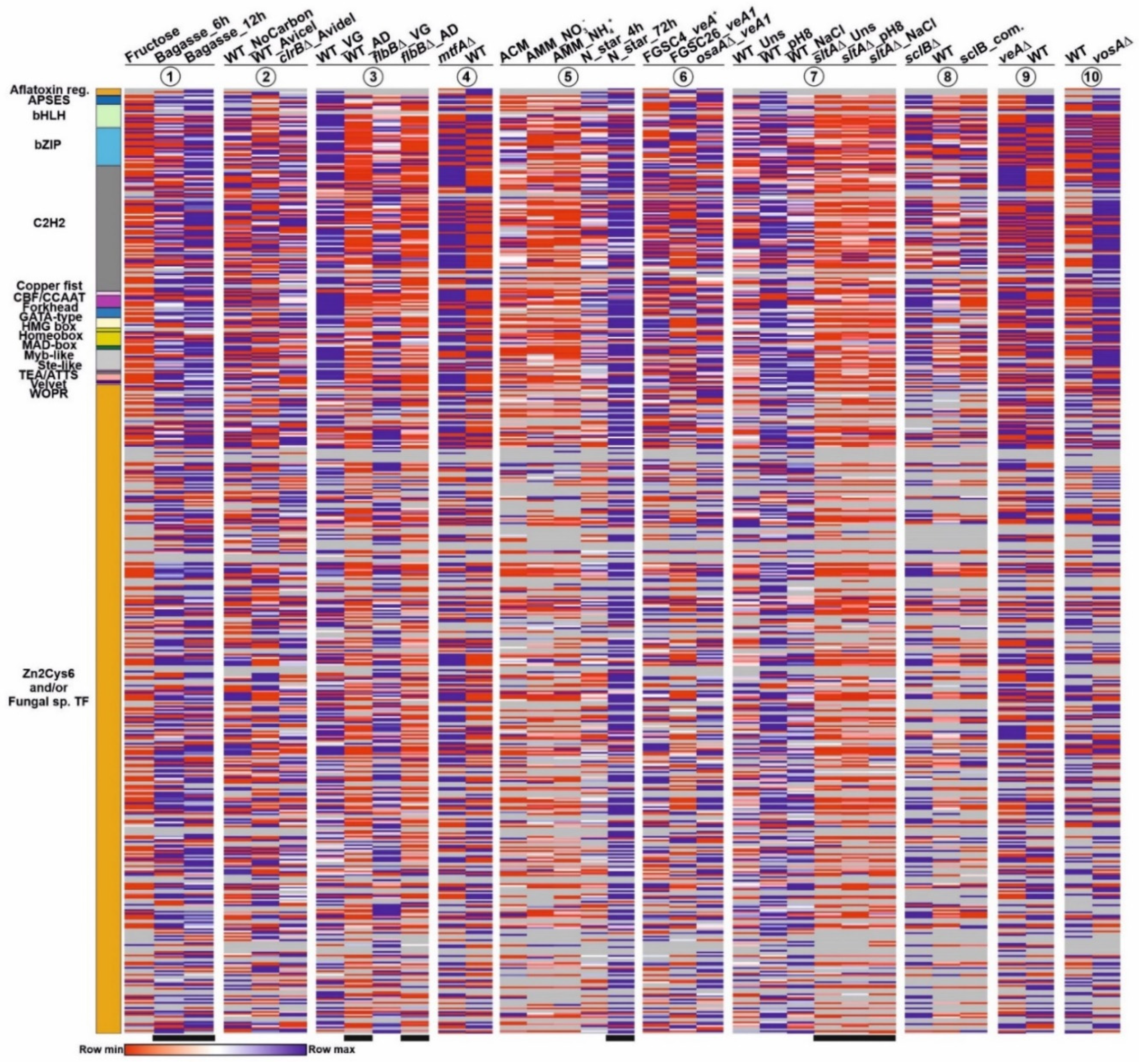
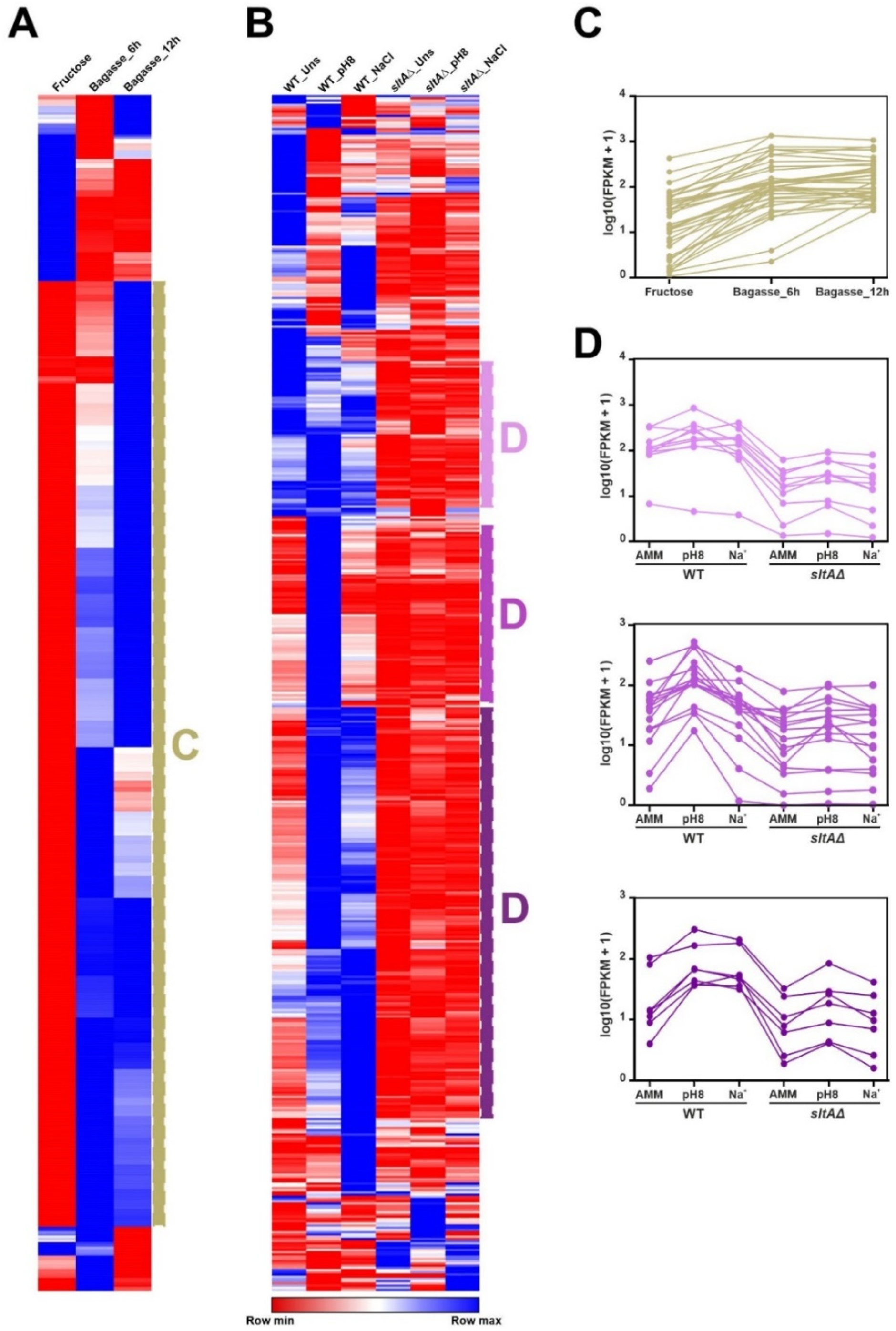
3.2. Discrepancy between Genomic Annotation and Transcriptomic Data
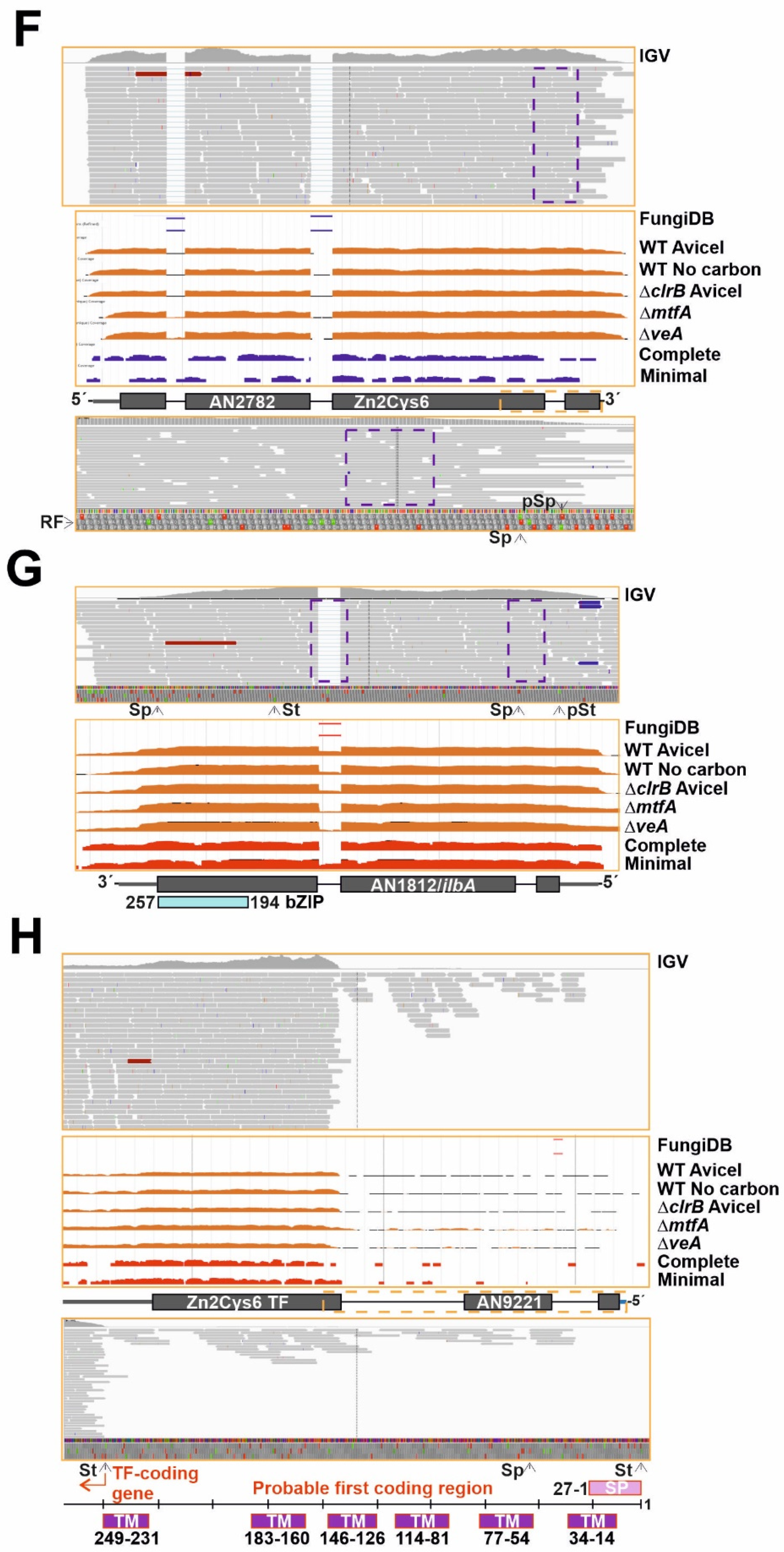
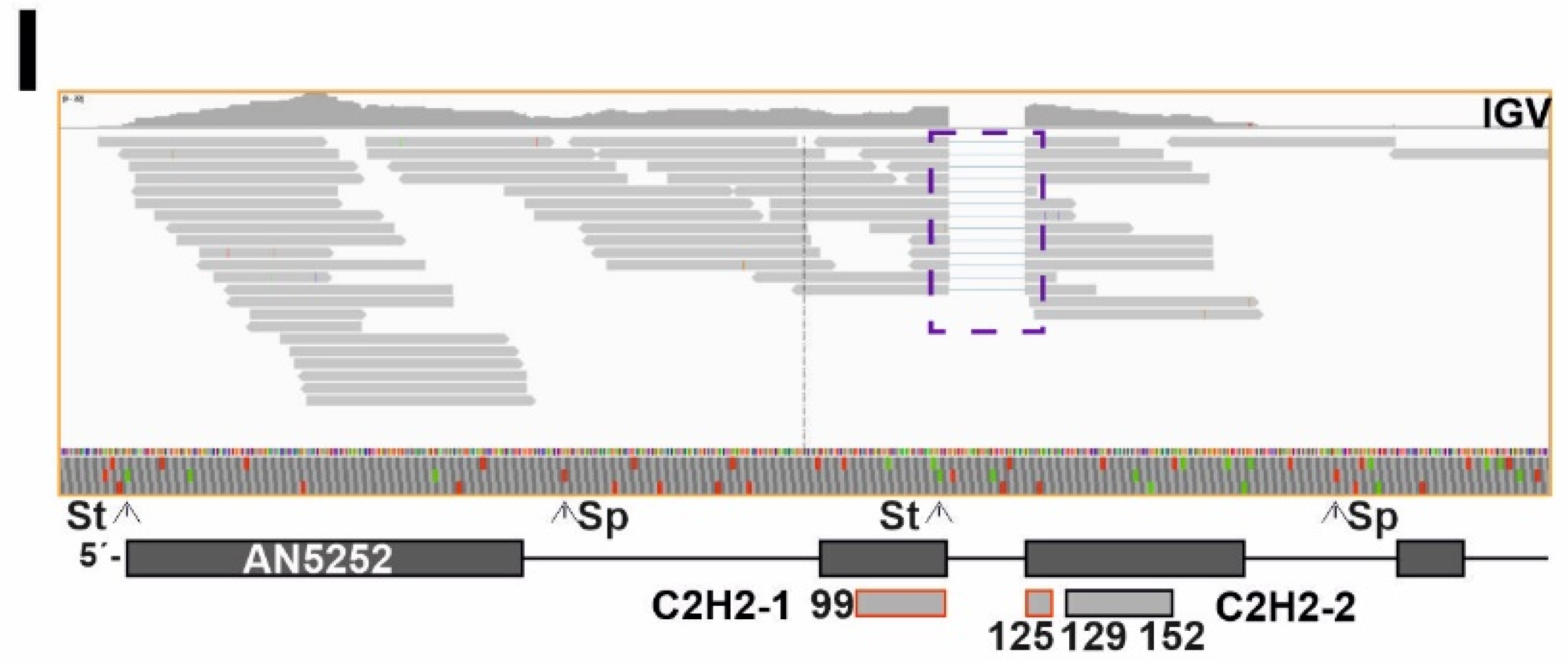
3.3. A Subset of Genes Potentially Coding for TFs, Mainly within the Binuclear Zinc Cluster Family, Remains Silent
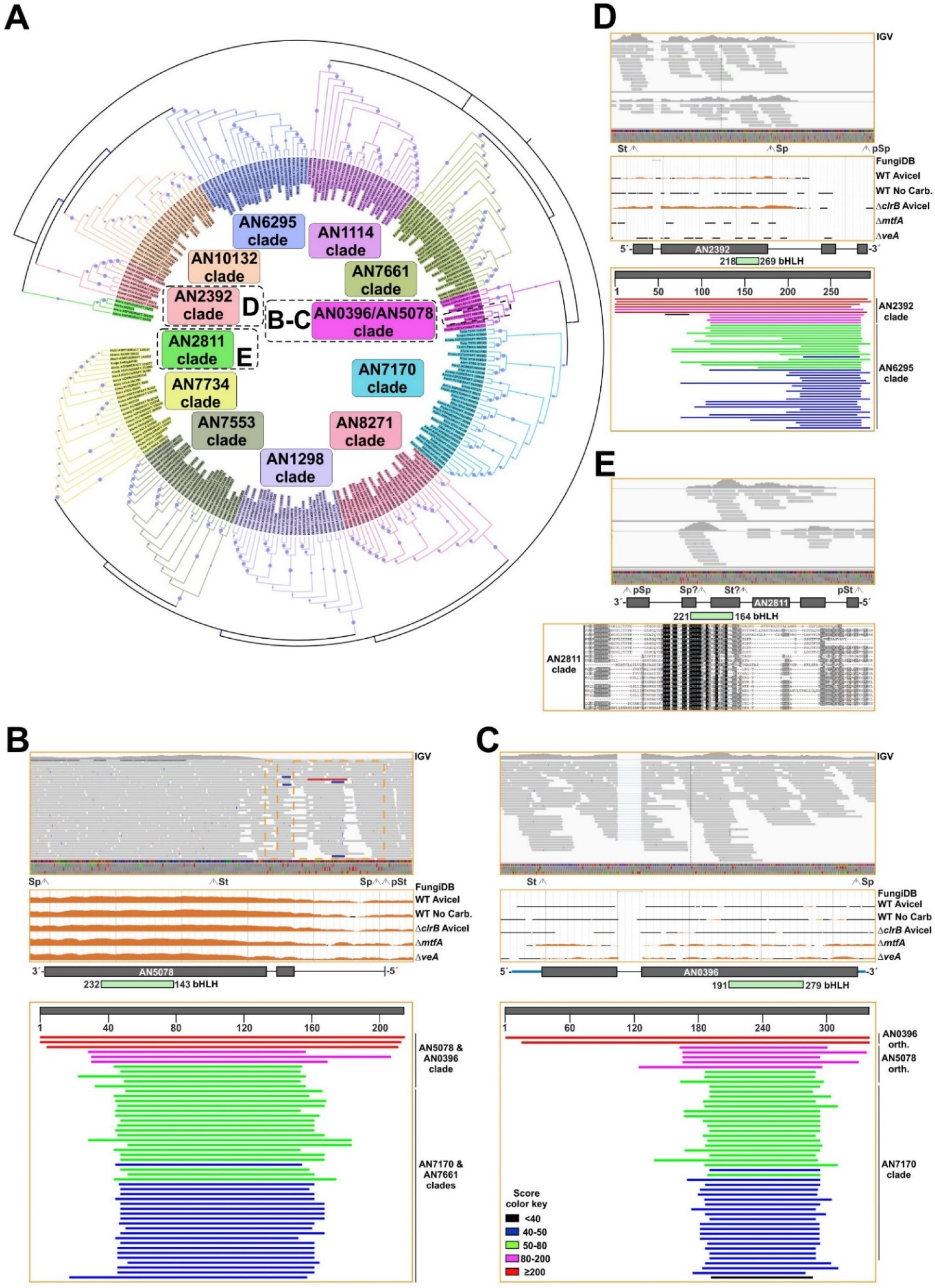
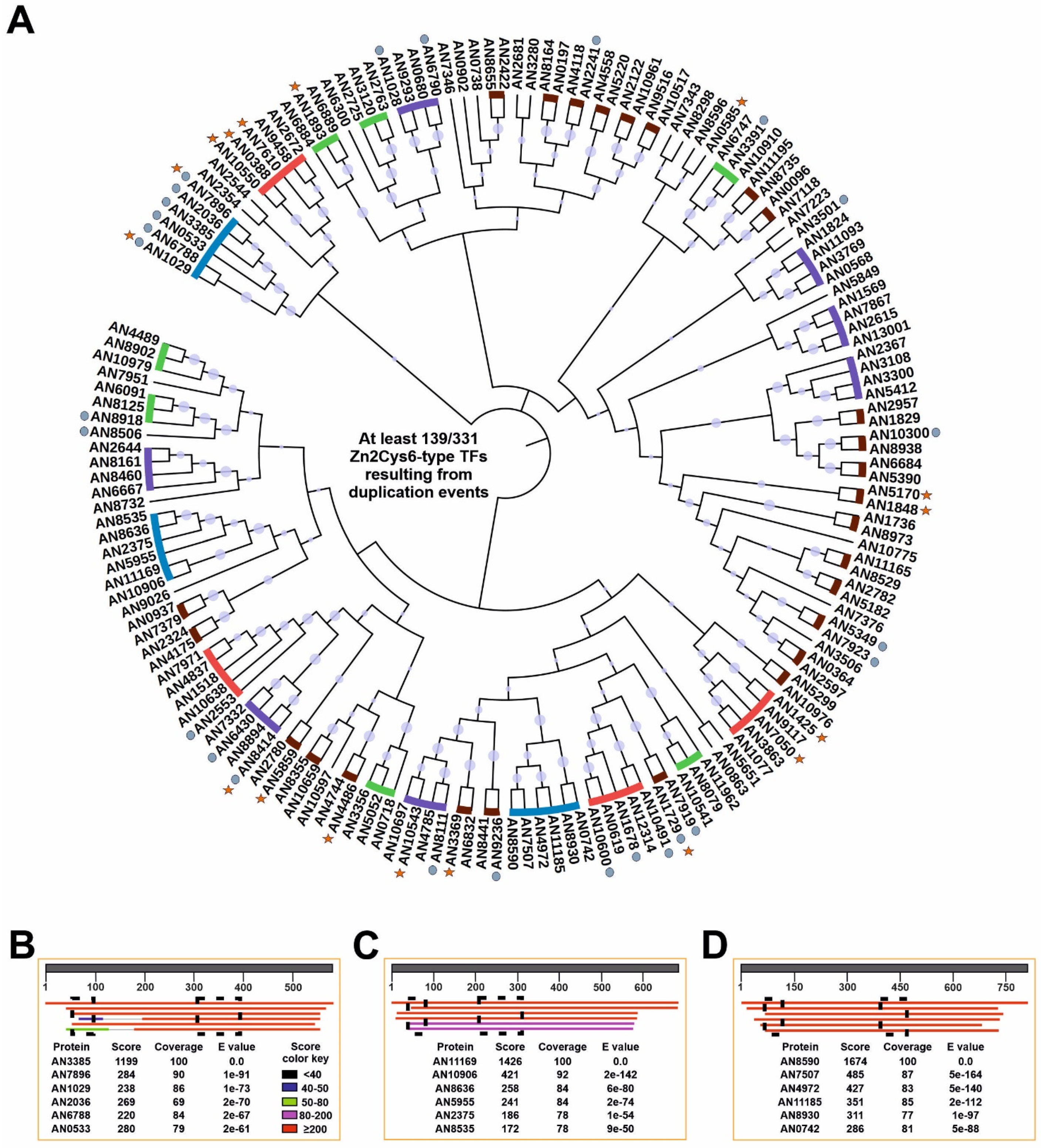
3.4. Duplication of TF-Coding Genes Impacts the Species Level
3.5. The Zn2Cys6 Family of TFs as a Paradigm of Gene Duplication Events Extending the Transcriptional Regulatory Potential of A. nidulans
3.6. Identification of a Zn2Cys6-Type Protein Necessary for Growth and Development
4. Discussion
How GRNs Evolve and What Fungi Can Teach Us about the Process
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, J.; Wang, H.H. Systematic and synthetic approaches to rewire regulatory networks. Curr. Opin. Syst. Biol. 2018, 8, 90–96. [Google Scholar] [CrossRef]
- Nocedal, I.; Johnson, A.D. How Transcription Networks Evolve and Produce Biological Novelty. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrells, T.R.; Johnson, A.D. Making Sense of Transcription Networks. Cell 2015, 161, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Voordeckers, K.; Pougach, K.; Verstrepen, K.J. How do regulatory networks evolve and expand throughout evolution? Curr. Opin. Biotechnol. 2015, 34, 180–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, C.S.; Sorrells, T.R.; Johnson, A.D. Protein-coding changes preceded cis-regulatory gains in a newly evolved transcription circuit. Science 2020, 367, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Spatafora, J.W.; Aime, M.C.; Grigoriev, I.V.; Martin, F.; Stajich, J.E.; Blackwell, M. The fungal tree of life: From molecular systematics to genome-scale phylogenies. Microbiol. Spectr. 2017, 5, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizsán, K.; Almási, É.; Merényi, Z.; Sahu, N.; Virágh, M.; Kószó, T.; Mondo, S.; Kiss, B.; Bálint, B.; Kües, U.; et al. Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity in fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 7409–7418. [Google Scholar] [CrossRef] [Green Version]
- Etxebeste, O.; Otamendi, A.; Garzia, A.; Espeso, E.A.; Cortese, M.S. Rewiring of transcriptional networks as a major event leading to the diversity of asexual multicellularity in fungi. Crit. Rev. Microbiol. 2019, 45, 548–563. [Google Scholar] [CrossRef]
- Kiss, E.; Hegedüs, B.; Varga, T.; Merényi, Z.; Kószó, T.; Bálint, B.; Prasanna, A.N.; Krizsán, K.; Riquelme, M.; Takeshita, N.; et al. Comparative genomics reveals the origin of fungal hyphae and multicellularity. Nat. Commun. 2019, 10, 4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, T.H.; Wieser, J.K.; Yu, J.-H. Asexual sporulation in Aspergillus nidulans. Microbiol. Mol. Biol. Rev. 1998, 62, 35–54. [Google Scholar] [CrossRef] [Green Version]
- Ojeda-López, M.; Chen, W.; Eagle, C.E.; Gutiérrez, G.; Jia, W.L.; Swilaiman, S.S.; Huang, Z.; Park, H.-S.; Yu, J.-H.; Cánovas, D.; et al. Evolution of asexual and sexual reproduction in the aspergilli. Stud. Mycol. 2018, 91, 37–59. [Google Scholar] [CrossRef]
- de Vries, R.P.; Riley, R.; Wiebenga, A.; Aguilar-Osorio, G.; Amillis, S.; Uchima, C.A.; Anderluh, G.; Asadollahi, M.; Askin, M.; Barry, K.; et al. Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 2017, 18, 28. [Google Scholar] [CrossRef] [Green Version]
- Wortman, J.R.; Gilsenan, J.M.; Joardar, V.; Deegan, J.; Clutterbuck, J.; Andersen, M.R.; Archer, D.; Bencina, M.; Braus, G.; Coutinho, P.; et al. The 2008 update of the Aspergillus nidulans genome annotation: A community effort. Fungal Genet. Biol. 2009, 46, S2–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basenko, E.; Pulman, J.; Shanmugasundram, A.; Harb, O.; Crouch, K.; Starns, D.; Warrenfeltz, S.; Aurrecoechea, C.; Stoeckert, C.; Kissinger, J.; et al. FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes. J. Fungi. 2018, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Coradetti, S.T.; Xiong, Y.; Glass, N.L. Analysis of a conserved cellulase transcriptional regulator reveals inducer-independent production of cellulolytic enzymes in Neurospora crassa. Microbiologyopen 2013, 2, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Sibthorp, C.; Wu, H.; Cowley, G.; Wong, P.W.H.; Palaima, P.; Morozov, I.Y.; Weedall, G.D.; Caddick, M.X. Transcriptome analysis of the filamentous fungus Aspergillus nidulans directed to the global identification of promoters. BMC Genom. 2013, 14, 847. [Google Scholar] [CrossRef] [Green Version]
- Lind, A.L.; Wisecaver, J.H.; Smith, T.D.; Feng, X.; Calvo, A.M.; Rokas, A. Examining the evolution of the regulatory circuit controlling secondary metabolism and development in the fungal genus Aspergillus. PLoS Genet. 2015, 11, e1005096. [Google Scholar] [CrossRef]
- Picazo, I.; Etxebeste, O.; Requena, E.; Garzia, A.; Espeso, E.A. Defining the transcriptional responses of Aspergillus nidulans to cation/alkaline pH stress and the role of the transcription factor SltA. Microb. Genom. 2020, 6, e000415. [Google Scholar] [CrossRef] [PubMed]
- Garzia, A.; Etxebeste, O.; Rodríguez-Romero, J.; Fischer, R.; Espeso, E.A.; Ugalde, U. Transcriptional changes in the transition from vegetative cells to asexual development in the model fungus Aspergillus nidulans. Eukaryot. Cell 2013, 12, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Oiartzabal-Arano, E.; Garzia, A.; Gorostidi, A.; Ugalde, U.; Espeso, E.A.; Etxebeste, O. Beyond asexual development: Modifications in the gene expression profile caused by the absence of the Aspergillus nidulans transcription factor FlbB. Genetics 2015, 199, 1127–1142. [Google Scholar] [CrossRef] [Green Version]
- Thieme, K.G.; Gerke, J.; Sasse, C.; Valerius, O.; Thieme, S.; Karimi, R.; Heinrich, A.K.; Finkernagel, F.; Smith, K.; Bode, H.B.; et al. Velvet domain protein VosA represses the zinc cluster transcription factor SclB regulatory network for Aspergillus nidulans asexual development, oxidative stress response and secondary metabolism. PLoS Genet. 2018, 14, e1007511. [Google Scholar] [CrossRef]
- Kim, M.-J.; Lee, M.-K.; Pham, H.Q.; Gu, M.J.; Zhu, B.; Son, S.-H.; Hahn, D.; Shin, J.-H.; Yu, J.-H.; Park, H.-S.; et al. The velvet regulator VosA governs survival and secondary metabolism of sexual spores in Aspergillus nidulans. Genes 2020, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Alkhayyat, F.; Ni, M.; Kim, S.C.; Yu, J.-H. The WOPR domain protein OsaA orchestrates development in Aspergillus nidulans. PLoS ONE 2015, 10, e0137554. [Google Scholar]
- Brown, N.A.; Ries, L.N.A.; Reis, T.F.; Rajendran, R.; Corrêa dos Santos, R.A.; Ramage, G.; Riaño-Pachón, D.M.; Goldman, G.H. RNAseq reveals hydrophobins that are involved in the adaptation of Aspergillus nidulans to lignocellulose. Biotechnol. Biofuels 2016, 9, 145. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Yang, L.; Ukil, L.; Osmani, A.; Nahm, F.; Davies, J.; De Souza, C.P.C.; Dou, X.; Perez-Balaguer, A.; Osmani, S.A. Rapid production of gene replacement constructs and generation of a green fluorescent protein-tagged centromeric marker in Aspergillus nidulans. Eukaryot. Cell 2004, 3, 1359–1362. [Google Scholar] [CrossRef] [Green Version]
- Prade, R.A.; Timberlake, W.E. The Aspergillus nidulans brlA regulatory locus consists of overlapping transcription units that are individually required for conidiophore development. EMBO J. 1993, 12, 2439–2447. [Google Scholar] [CrossRef]
- Nayak, T.; Szewczyk, E.; Oakley, C.E.; Osmani, A.; Ukil, L.; Murray, S.L.; Hynes, M.J.; Osmani, S.A.; Oakley, B.R. A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics 2006, 172, 1557–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilburn, J.; Scazzocchio, C.; Taylor, G.G.; Zabicky-Zissman, J.H.; Lockington, R.A.; Davies, R.W. Transformation by integration in Aspergillus nidulans. Gene 1983, 26, 205–221. [Google Scholar] [CrossRef]
- Szewczyk, E.; Nayak, T.; Oakley, C.E.; Edgerton, H.; Xiong, Y.; Taheri-Talesh, N.; Osmani, S.A.; Oakley, B.R. Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 2007, 1, 3111–3120. [Google Scholar] [CrossRef] [PubMed]
- Garzia, A.; Etxebeste, O.; Herrero-Garcia, E.; Fischer, R.; Espeso, E.A.; Ugalde, U. Aspergillus nidulans FlbE is an upstream developmental activator of conidiation functionally associated with the putative transcription factor FlbB. Mol. Microbiol. 2009, 71, 172–184. [Google Scholar] [CrossRef]
- Etxebeste, O.; Espeso, E.A. Aspergillus nidulans in the post-genomic era: A top-model filamentous fungus for the study of signaling and homeostasis mechanisms. Int. Microbiol. 2020, 23, 5–22. [Google Scholar] [CrossRef]
- Shelest, E. Transcription Factors in fungi: TFome dynamics, three major families, and dual-specificity TFs. Front. Genet. 2017, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Todd, R.B.; Zhou, M.; Ohm, R.A.; Leeggangers, H.A.C.F.; Visser, L.; de Vries, R.P. Prevalence of transcription factors in ascomycete and basidiomycete fungi. BMC Genom. 2014, 15, 214. [Google Scholar] [CrossRef] [Green Version]
- Bat-Ochir, C.; Kwak, J.-Y.; Koh, S.-K.; Jeon, M.-H.; Chung, D.; Lee, Y.-W.; Chae, S.-K. The signal peptide peptidase SppA is involved in sterol regulatory element-binding protein cleavage and hypoxia adaptation in Aspergillus nidulans. Mol. Microbiol. 2016, 100, 635–655. [Google Scholar] [CrossRef] [Green Version]
- Salzberg, S.L. Next-generation genome annotation: We still struggle to get it right. Genome Biol. 2019, 20, 92. [Google Scholar] [CrossRef] [Green Version]
- Vallim, M.A.; Miller, K.Y.; Miller, B.L. Aspergillus SteA (Sterile12-like) is a homeodomain-C2/H2-Zn+2 finger transcription factor required for sexual reproduction. Mol. Microbiol. 2002, 36, 290–301. [Google Scholar] [CrossRef]
- Strittmatter, A.W.; Irniger, S.; Braus, G.H. Induction of jlbA mRNA synthesis for a putative bZIP protein of Aspergillus nidulans by amino acid starvation. Curr. Genet. 2001, 39, 327–334. [Google Scholar] [CrossRef]
- Grau, M.F.; Entwistle, R.; Chiang, Y.-M.; Ahuja, M.; Oakley, C.E.; Akashi, T.; Wang, C.C.C.; Todd, R.B.; Oakley, B.R. Hybrid transcription factor engineering activates the silent secondary metabolite gene cluster for (+)-Asperlin in Aspergillus nidulans. ACS Chem. Biol. 2018, 13, 3193–3205. [Google Scholar] [CrossRef]
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C.J., Jr. Using OrthoMCL to Assign Proteins to OrthoMCL-DB Groups or to Cluster Proteomes Into New Ortholog Groups. Curr. Protoc. Bioinforma. 2011, 35, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Arst, H.N.; Hondmann, D.H.A.; Visser, J. A translocation activating the cryptic nitrogen regulation gene areB inactivates a previously unidentified gene involved in glycerol utilisation in Aspergillus nidulans. Mol. Gen. Genet. MGG 1990, 223, 134–137. [Google Scholar] [CrossRef]
- Tüncher, A.; Reinke, H.; Martic, G.; Caruso Maria, L.; Axe lA., B. A basic-region helix–loop–helix protein-encoding gene (devR) involved in the development of Aspergillus nidulans. Mol. Microbiol. 2004, 52, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Caruso, M.L.; Litzka, O.; Martic, G.; Lottspeich, F.; Brakhage, A.A. Novel Basic-region Helix–Loop–Helix transcription factor (AnBH1) of Aspergillus nidulans counteracts the CCAAT-binding complex AnCF in the promoter of a penicillin biosynthesis gene. J. Mol. Biol. 2002, 323, 425–439. [Google Scholar] [CrossRef]
- Caddick, M.X.; Brownlee, A.G.; Arst, H.N. Phosphatase regulation in Aspergillus nidulans: Responses to nutritional starvation. Genet. Res. 1986, 47, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.S.; Lohmar, M.J.; Ahmed, S.; Etxebeste, O.; Espeso, A.E.; Calvo, M.A. UrdA controls secondary metabolite production and the balance between asexual and sexual development in Aspergillus nidulans. Genes 2018, 9, 570. [Google Scholar] [CrossRef] [Green Version]
- Nützmann, H.-W.; Scazzocchio, C.; Osbourn, A. Metabolic Gene Clusters in Eukaryotes. Annu. Rev. Genet. 2018, 52, 159–183. [Google Scholar] [CrossRef]
- Rokas, A.; Wisecaver, J.H.; Lind, A.L. The birth, evolution and death of metabolic gene clusters in fungi. Nat. Rev. Microbiol. 2018. [Google Scholar] [CrossRef]
- Todd, R.B.; Andrianopoulos, A. Evolution of a fungal regulatory gene family: The Zn(II)2Cys6 binuclear cluster DNA binding motif. Fungal Genet. Biol. 1997, 21, 388–405. [Google Scholar] [CrossRef]
- Chiang, Y.-M.; Szewczyk, E.; Davidson, A.D.; Keller, N.; Oakley, B.R.; Wang, C.C.C. A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, Asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 2009, 131, 2965–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boylan, M.T.; Mirabito, P.M.; Willett, C.E.; Zimmerman, C.R.; Timberlake, W.E. Isolation and physical characterization of three essential conidiation genes from Aspergillus nidulans. Mol. Cell. Biol. 1987, 7, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Siggers, T.; Reddy, J.; Barron, B.; Bulyk, M.L. Diversification of Transcription Factor Paralogs via Noncanonical Modularity in C2H2 Zinc Finger DNA Binding. Mol. Cell 2014, 55, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Verd, B.; Monk, N.A.M.; Jaeger, J. Modularity, criticality, and evolvability of a developmental gene regulatory network. eLife 2019, 8, e42832. [Google Scholar] [CrossRef]
- Lee, M.-K.; Kwon, N.-J.; Choi, J.M.; Lee, I.-S.; Jung, S.; Yu, J.-H. NsdD is a key repressor of asexual development in Aspergillus nidulans. Genetics 2014, 197, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-K.; Kwon, N.-J.; Lee, I.-S.; Jung, S.; Kim, S.-C.; Yu, J.-H. Negative regulation and developmental competence in Aspergillus. Sci. Rep. 2016, 6, 28874. [Google Scholar] [CrossRef] [Green Version]
- Skromne, I.; Sanchez, O.; Aguirre, J. Starvation stress modulates the expression of the Aspergillus nidulans brlA regulatory gene. Microbiology 1995, 141, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerqueira, G.C.; Arnaud, M.B.; Inglis, D.O.; Skrzypek, M.S.; Binkley, G.; Simison, M.; Miyasato, S.R.; Binkley, J.; Orvis, J.; Shah, P.; et al. The Aspergillus Genome Database: Multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res. 2014, 42, D705–D710. [Google Scholar] [CrossRef] [Green Version]
- Kwon, N.-J.; Garzia, A.; Espeso, E.A.; Ugalde, U.; Yu, J.-H. FlbC is a putative nuclear C2H2 transcription factor regulating development in Aspergillus nidulans. Mol. Microbiol. 2010, 77, 1203–1219. [Google Scholar] [CrossRef]
- Garzia, A.; Etxebeste, O.; Herrero-García, E.; Ugalde, U.; Espeso, E.A. The concerted action of bZip and cMyb transcription factors FlbB and FlbD induces brlA expression and asexual development in Aspergillus nidulans. Mol. Microbiol. 2010, 75, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Son, S.-H.; Son, Y.-E.; Cho, H.-J.; Chen, W.; Lee, M.-K.; Kim, L.-H.; Han, D.-M.; Park, H.-S. Homeobox proteins are essential for fungal differentiation and secondary metabolism in Aspergillus nidulans. Sci. Rep. 2020, 10, 6094. [Google Scholar] [CrossRef] [Green Version]
- Marshall, M.A.; Timberlake, W.E. Aspergillus nidulans wetA activates spore-specific gene expression. Mol. Cell. Biol. 1991, 11, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.Y.; Wu, J.; Miller, B.L. StuA is required for cell pattern formation in Aspergillus. Genes Dev. 1992, 6, 1770–1782. [Google Scholar] [CrossRef] [Green Version]
- Busby, T.M.; Miller, K.Y.; Miller, B.L. Suppression and enhancement of the Aspergillus nidulans medusa mutation by altered dosage of the bristle and stunted Genes. Genetics 1996, 143, 155–163. [Google Scholar] [CrossRef]
- Igler, C.; Lagator, M.; Tkačik, G.; Bollback, J.P.; Guet, C.C. Evolutionary potential of transcription factors for gene regulatory rewiring. Nat. Ecol. Evol. 2018, 2, 1633–1643. [Google Scholar] [CrossRef]
- Noman, N.; Monjo, T.; Moscato, P.; Iba, H. Evolving robust gene regulatory networks. PLoS ONE 2015, 10, e0116258. [Google Scholar] [CrossRef] [Green Version]
- Kitano, H. Biological robustness. Nat. Rev. Genet. 2004, 5, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Steenwyk, J.L.; Chang, Y.; Wang, Y.; James, T.Y.; Stajich, J.E.; Spatafora, J.W.; Groenewald, M.; Dunn, C.W.; Hittinger, C.T.; et al. A genome-scale phylogeny of the kingdom Fungi. Curr. Biol. 2021, 31, 1653–1665. [Google Scholar] [CrossRef]
- de Mendoza, A.; Sebé-Pedrós, A.; Šestak, M.S.; Matejčić, M.; Torruella, G.; Domazet-Lošo, T.; Ruiz-Trillo, I. Transcription factor evolution in eukaryotes and the assembly of the regulatory toolkit in multicellular lineages. Proc. Natl. Acad. Sci. USA 2013, 110, E4858–E4866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Mendoza, A.; Sebé-Pedrós, A. Origin and evolution of eukaryotic transcription factors. Curr. Opin. Genet. Dev. 2019, 58–59, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Mead, M.E.; Borowsky, A.T.; Joehnk, B.; Steenwyk, J.L.; Shen, X.-X.; Sil, A.; Rokas, A. Recurrent loss of abaA, a master regulator of asexual development in filamentous fungi, correlates with changes in genomic and morphological Traits. Genome Biol. Evol. 2020, 12, 1119–1130. [Google Scholar] [CrossRef]
- Pires, N.; Dolan, L. Early evolution of bHLH proteins in plants. Plant Signal. Behav. 2010, 5, 911–912. [Google Scholar] [CrossRef]
- Pires, N.; Dolan, L. Origin and diversification of Basic-Helix-Loop-Helix proteins in plants. Mol. Biol. Evol. 2010, 27, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Xu, F.; Shimeld, S.M. Phylogenetics of lophotrochozoan bHLH genes and the evolution of lineage-specific gene duplicates. Genome Biol. Evol. 2017, 9, 869–886. [Google Scholar] [CrossRef] [Green Version]
- Lallemand, T.; Leduc, M.; Landès, C.; Rizzon, C.; Lerat, E. An overview of duplicated gene detection methods: Why the duplication mechanism has to be accounted for in their choice. Genes 2020, 11, 1046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Corrochano, L.M.; Kuo, A.; Marcet-Houben, M.; Polaino, S.; Salamov, A.; Villalobos-Escobedo, J.M.; Grimwood, J.; Álvarez, M.I.; Avalos, J.; Bauer, D.; et al. Expansion of Signal Transduction Pathways in Fungi by Extensive Genome Duplication. Curr. Biol. 2016, 26, 1577–1584. [Google Scholar] [CrossRef] [Green Version]
- Khaldi, N.; Wolfe, K.H. Elusive origins of the extra genes in Aspergillus oryzae. PLoS ONE 2008, 3, e3036. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.A.; Yavor, A.M.; Massa, H.F.; Trask, B.J.; Eichler, E.E. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 2001, 11, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerke, J.; Braus, G.H. Manipulation of fungal development as source of novel secondary metabolites for biotechnology. Appl. Microbiol. Biotechnol. 2014, 98, 8443–8455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.; Ekstrom, A.; Li, X.; Yin, Y. HGT-Finder: A new tool for horizontal gene transfer finding and application to Aspergillus genomes. Toxins 2015, 7, 4035–4053. [Google Scholar] [CrossRef] [Green Version]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef]
- Tamayo, E.N.; Villanueva, A.; Hasper, A.A.; de Graaff, L.H.; Ramón, D.; Orejas, M. CreA mediates repression of the regulatory gene xlnR which controls the production of xylanolytic enzymes in Aspergillus nidulans. Fungal Genet. Biol. 2008, 45, 984–993. [Google Scholar] [CrossRef]
- Kowalczyk, J.E.; Gruben, B.S.; Battaglia, E.; Wiebenga, A.; Majoor, E.; de Vries, R.P. Genetic interaction of Aspergillus nidulans galR, xlnR and araR in regulating D-galactose and L-arabinose release and catabolism gene expression. PLoS ONE 2015, 10, e0143200. [Google Scholar] [CrossRef]
- Vienken, K.; Scherer, M.; Fischer, R. The Zn(II)2Cys6 putative Aspergillus nidulans transcription factor repressor of sexual development inhibits sexual development under low-carbon conditions and in submersed culture. Genetics 2005, 169, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vienken, K.; Fischer, R. The Zn(II)2Cys6 putative transcription factor NosA controls fruiting body formation in Aspergillus nidulans. Mol. Microbiol. 2006, 61, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Hynes, M.J.; Murray, S.L.; Duncan, A.; Khew, G.S.; Davis, M.A. Regulatory genes controlling fatty acid catabolism and peroxisomal functions in the filamentous fungus Aspergillus nidulans. Eukaryot. Cell 2006, 5, 794–805. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Timberlake, W.E. Identification of Aspergillus brlA response elements (BREs) by genetic selection in yeast. Genetics 1993, 133, 29–38. [Google Scholar] [CrossRef]
- Dyer, P.S.; O’Gorman, C.M. Sexual development and cryptic sexuality in fungi: Insights from Aspergillus species. FEMS Microbiol. Rev. 2012, 36, 165–192. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etxebeste, O. Transcription Factors in the Fungus Aspergillus nidulans: Markers of Genetic Innovation, Network Rewiring and Conflict between Genomics and Transcriptomics. J. Fungi 2021, 7, 600. https://doi.org/10.3390/jof7080600
Etxebeste O. Transcription Factors in the Fungus Aspergillus nidulans: Markers of Genetic Innovation, Network Rewiring and Conflict between Genomics and Transcriptomics. Journal of Fungi. 2021; 7(8):600. https://doi.org/10.3390/jof7080600
Chicago/Turabian StyleEtxebeste, Oier. 2021. "Transcription Factors in the Fungus Aspergillus nidulans: Markers of Genetic Innovation, Network Rewiring and Conflict between Genomics and Transcriptomics" Journal of Fungi 7, no. 8: 600. https://doi.org/10.3390/jof7080600