
Fungal Endophytic Community and Diversity Associated with Desert Shrubs Driven by Plant Identity and Organ Differentiation in Extremely Arid Desert Ecosystem


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Sampling
2.2. Molecular Analysis
2.3. Bioinformatics Processing
2.4. Statistical Analysis
3. Results
3.1. Characterization of Illumina Sequencing Data
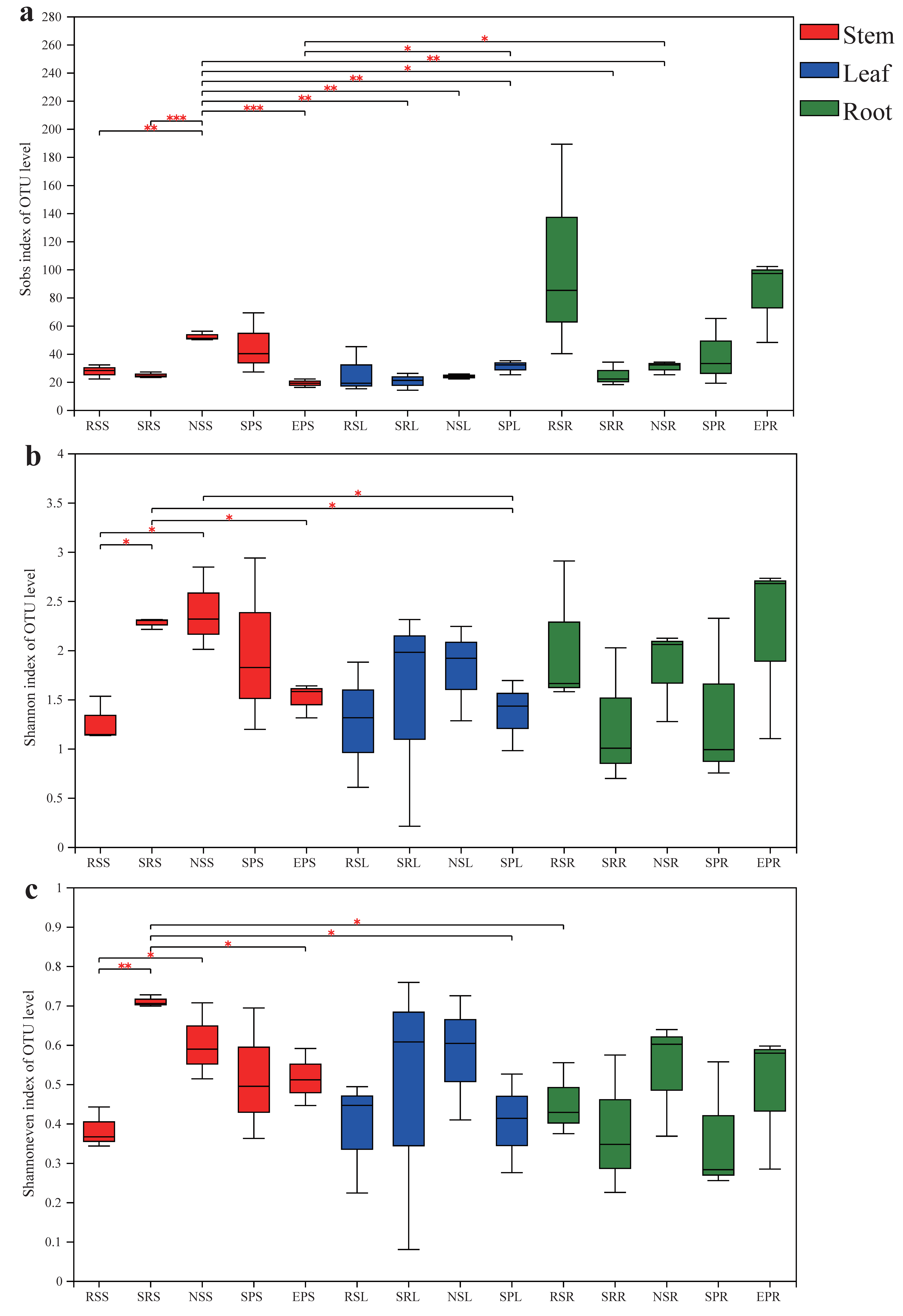
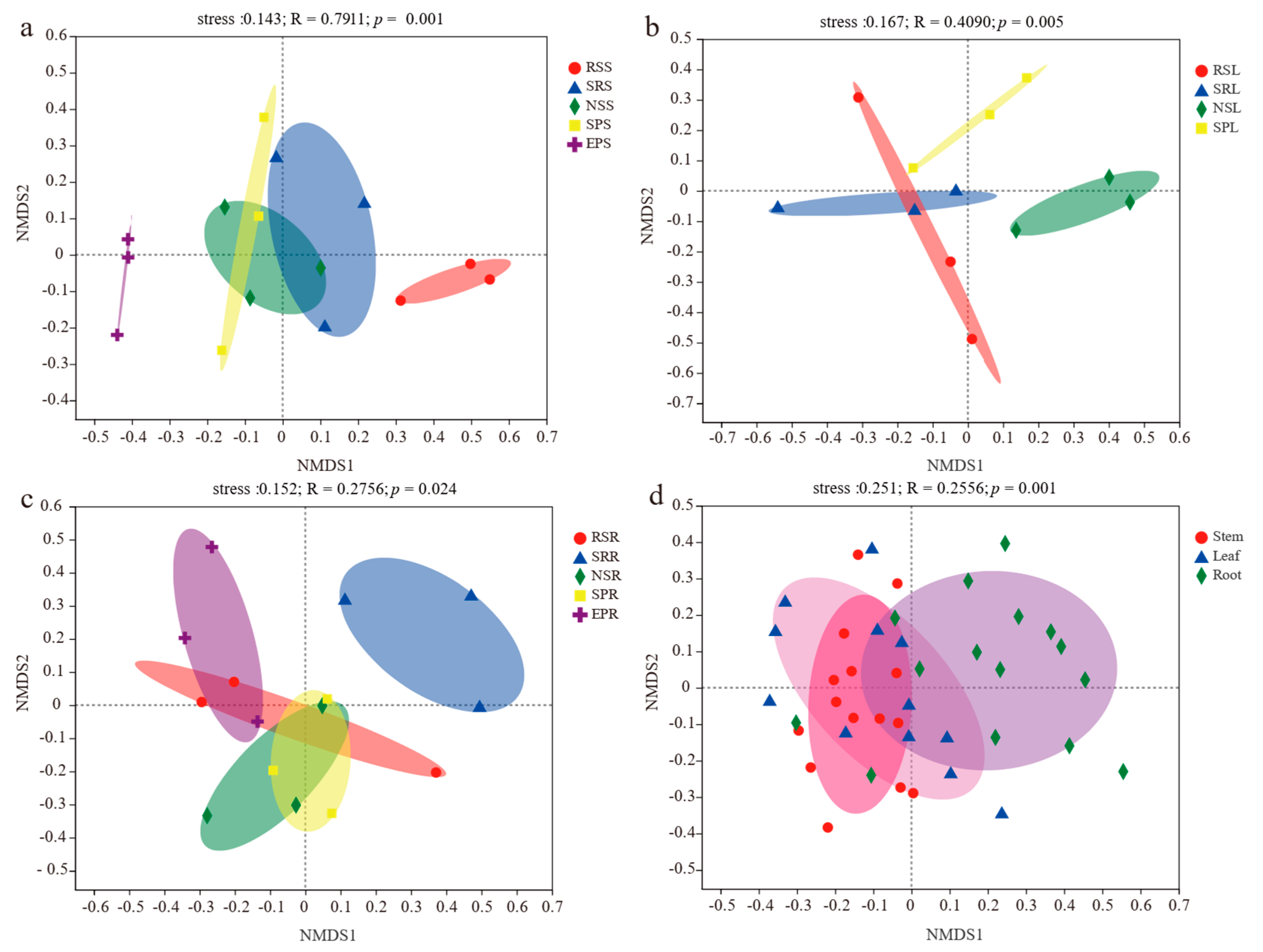
3.2. Alpha and Beta Diversities
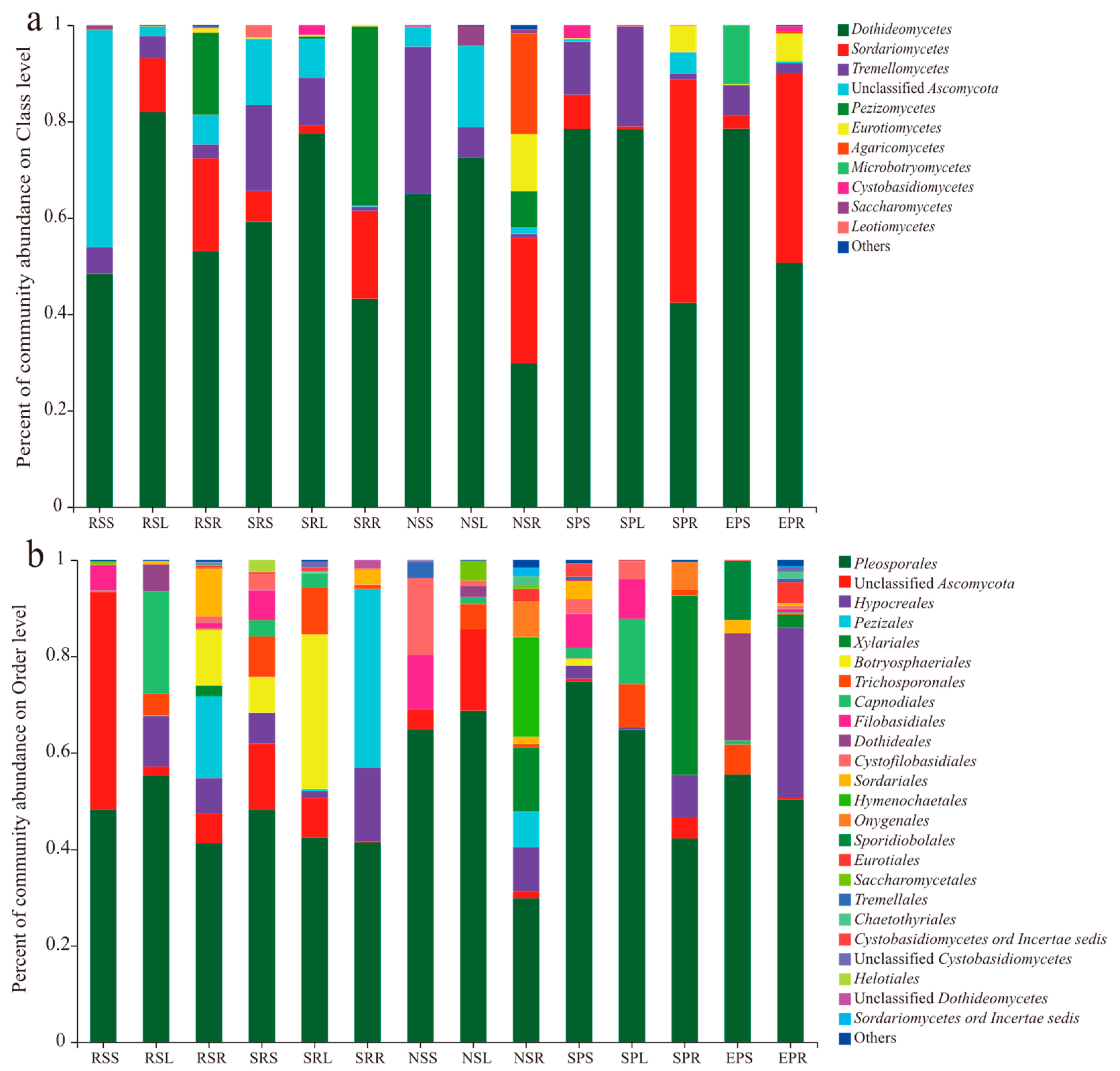
3.3. Community Composition of Endophytic Fungi Across Hosts and Organ Compartments
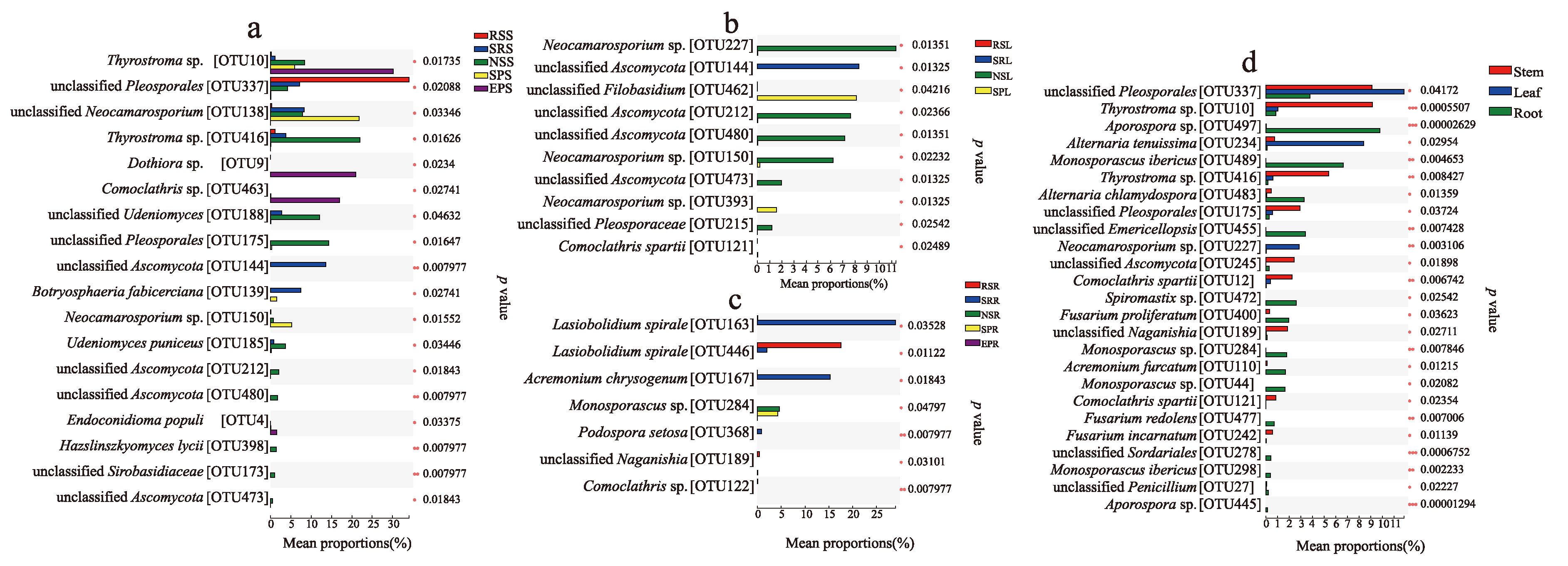
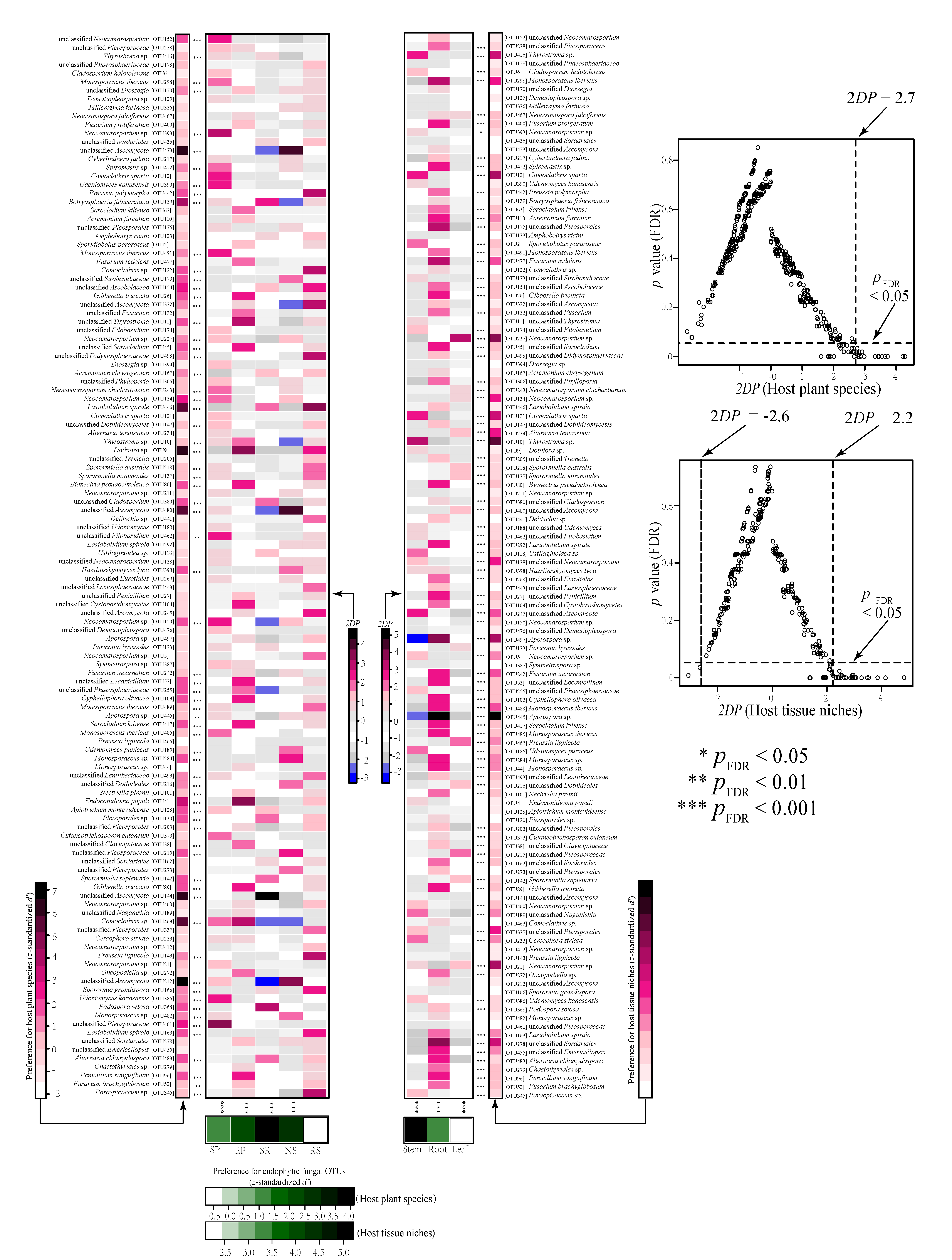
3.4. Host and Organ Preferences of Endophytic Fungi
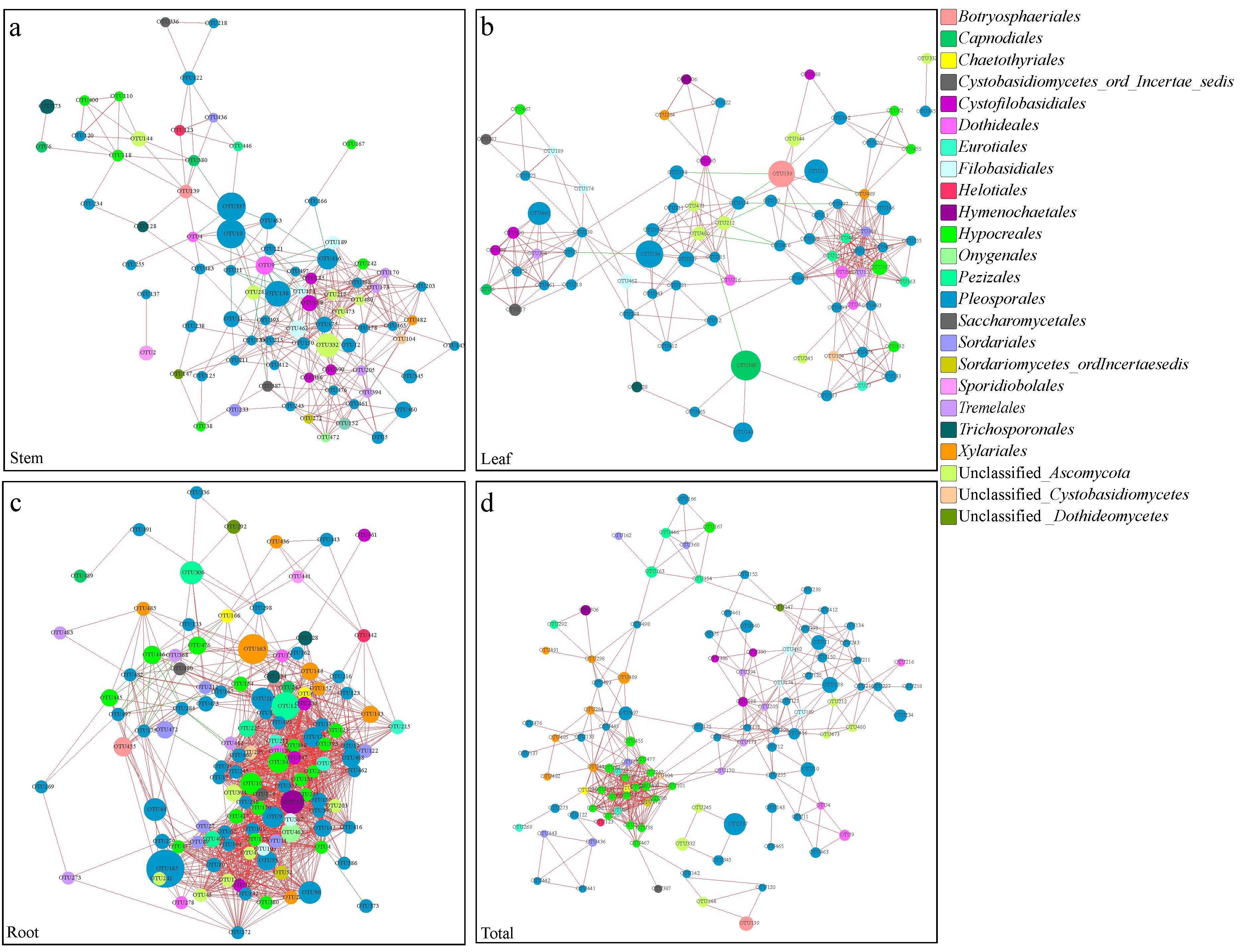
3.5. Co-Occurrence Networks of Endophytic Fungi
4. Discussion
4.1. Fungal Community Associated with Xerophytic Desert Shrubs
4.2. Host Selection Shape Fungal Assembly in Each Compartment Niches
4.3. Organ Differentiation and Tissue-Specificity of Endophytic Fungi
4.4. Co-Occurrence Network and Keystone Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuo, Y.L.; Su, F.; He, X.L.; Li, M. Colonization by dark septate endophytes improves the growth of Hedysarum scoparium under multiple inoculum levels. Symbiosis 2020, 82, 201–214. [Google Scholar] [CrossRef]
- Shi, Y.; Yan, X.; Zhao, P.; Yin, H.; Zhao, X.; Xiao, H.L.; Li, X.R.; Chen, G.X.; Ma, X.F. Transcriptomic analysis of a tertiary relict plant, extreme xerophyte Reaumuria soongorica to identify genes related to drought adaptation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Zhang, C.; Li, X.; Wu, H.; Wang, P.; Wang, Y.; Wu, X.C.; Li, W.; Huang, Y.M. Differences in water-use strategies along an aridity gradient between two coexisting desert shrubs (Reaumuria soongorica and Nitraria sphaerocarpa): Isotopic approaches with physiological evidence. Plant Soil 2017, 419, 169–187. [Google Scholar] [CrossRef]
- Beckers, B.; Op De Beeck, M.; Weyens, N.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Hassani, M.A.; Duran, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Sangamesh, M.B.; Jambagi, S.; Vasanthakumari, M.M.; Shetty, N.J.; Kolte, H.; Ravikanth, G.; Nataraja, K.N.; Shaanker, R.U. Thermotolerance of fungal endophytes isolated from plants adapted to the Thar Desert, India. Symbiosis 2018, 75, 135–147. [Google Scholar] [CrossRef]
- Hosseyni Moghaddam, M.S.; Safaie, N.; Soltani, J.; Hagh-Doust, N. Desert-adapted fungal endophytes induce salinity and drought stress resistance in model crops. Plant Physiol. Biochem. 2021, 160, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Massimo, N.C.; Nandi Devan, M.M.; Arendt, K.R.; Wilch, M.H.; Riddle, J.M.; Furr, S.H.; Arnold, A.E. Fungal endophytes in aboveground tissues of desert plants: Infrequent in culture, but highly diverse and distinctive symbionts. Microb. Ecol. 2015, 70, 61–76. [Google Scholar] [CrossRef]
- Ali, A.H.; Radwan, U.; El-Zayat, S.; El-Sayed, M.A. Desert plant-fungal endophytic association: The beneficial aspects to their hosts. Biol. Forum Int. J. 2018, 10, 138–145. [Google Scholar]
- Martin, F.M.; Uroz, S.; Barker, D.G. Ancestral alliances: Plant mutualistic symbioses with fungi and bacteria. Science 2017, 356, eaad4501. [Google Scholar] [CrossRef] [PubMed]
- Hacquard, S.; Garrido-Oter, R.; Gonzalez, A.; Spaepen, S.; Ackermann, G.; Lebeis, S.; McHardy, A.C.; Dangl, J.L.; Knight, R.; Ley, R.; et al. Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microb. 2015, 17, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, C.; Zhu, Y.G.; Wang, J.T.; Singh, B.; Han, L.L.; Shen, J.P.; Li, P.P.; Wang, G.B.; Wu, C.F.; Ge, A.H.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2020, 229, 1091–1104. [Google Scholar] [CrossRef]
- Cordovez, V.; Dini-Andreote, F.; Carrión, V.J.; Raaijmakers, J.M. Ecology and evolution of plant microbiomes. Annu. Rev. Microbiol. 2019, 73, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Martínez-García, L.B.; Armas, C.; Miranda, J.; Padilla, F.M.; Pugnaire, F.I. Shrubs influence arbuscular mycorrhizal fungi communities in a semi-arid environment. Soil Biol. Biochem. 2011, 43, 682–689. [Google Scholar] [CrossRef]
- Bonito, G.; Reynolds, H.; Robeson, M.S.; Nelson, J.; Hodkinson, B.P.; Tuskan, G.; Vilgalys, R. Plant host and soil origin influence fungal and bacterial assemblages in the roots of woody plants. Mol. Ecol. 2014, 23, 3356–3370. [Google Scholar] [CrossRef]
- Durand, A.; Maillard, F.; Foulon, J.; Gweon, H.S.; Valot, B.; Chalot, M. Environmental metabarcoding reveals contrasting belowground and aboveground fungal communities from Poplar at a Hg phytomanagement Site. Mol. Ecol. 2017, 74, 795–809. [Google Scholar] [CrossRef]
- Dastogeer, K.M.; Li, H.; Sivasithamparam, K.; Jones, M.G.K.; Wylie, S.J. Host specificity of endophytic mycobiota of wild Nicotiana plants from arid regions of northern Australia. Microb. Ecol. 2018, 75, 74–87. [Google Scholar] [CrossRef]
- Coleman-Derr, D.; Desgarennes, D.; Fonseca-Garcia, C.; Gross, S.; Clingenpeel, S.; Woyke, T.; North, G.; Visel, A.; Partida-Martinez, L.P.; Tringe, S.G. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 2016, 209, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Fonseca-García, C.; Coleman-Derr, D.; Garrido, E.; Visel, A.; Tringe, S.G.; Partida-Martínez, L.P. The cacti microbiome: Interplay between habitat-filtering and host-specificity. Front. Microbiol. 2016, 7, 150. [Google Scholar] [CrossRef]
- Jones, D.L.; Nguyen, C.; Finlay, R.D. Carbon flow in the rhizosphere: Carbon trading at the soil–root interface. Plant Soil 2009, 321, 5–33. [Google Scholar] [CrossRef]
- Del Pilar Martínez-Diz, M.; Andrés-Sodupe, M.; Bujanda, R.; Díaz-Losada, E.; Eichmeier, A.; Gramaje, D. Soil-plant compartments affect fungal microbiome diversity and composition in grapevine. Fungal Ecol. 2019, 41, 234–244. [Google Scholar] [CrossRef]
- Qian, X.; Li, H.; Wang, Y.; Wu, B.; Wu, M.; Chen, L.; Li, X.C.; Zhang, Y.; Wang, X.P.; Shi, M.M.; et al. Leaf and root endospheres harbor lower fungal diversity and less Complex fungal co-occurrence patterns than rhizosphere. Front. Microbiol. 2019, 10, 1015. [Google Scholar] [CrossRef]
- Toju, H.; Kurokawa, H.; Kenta, T. Factors influencing leaf- and root-associated communities of bacteria and fungi across 33 plant orders in a grassland. Front. Microbiol. 2019, 10, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara-Araya, M.J.; Vilo, C.; Urzúa, A.; González-Teuber, M. Differences in community composition of endophytic fungi between above- and below-ground tissues of Aristolochia chilensis in an arid ecosystem. Rev. Chil. Hist. Nat. 2020, 93, 3. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Q.; Lu, X.D.; Okane, L.; Kakishima, M. Endophytic fungal community in stems and leaves of plants from desert areas in China. Mycol. Progress 2012, 11, 781–790. [Google Scholar] [CrossRef]
- Li, J.L.; Sun, X.; Zheng, Y.; Lü, P.P.; Wang, Y.L.; Guo, L.D. Diversity and community of culturable endophytic fungi from stems and roots of desert halophytes in northwest China. MycoKeys 2020, 62, 75–95. [Google Scholar] [CrossRef]
- Del Olmo-Ruiz, M.; Arnold, A.E. Interannual variation and host affiliations of endophytic fungi associated with ferns at La Selva, Costa Rica. Mycologia 2014, 106, 8–21. [Google Scholar] [CrossRef]
- Sandberg, D.C.; Battista, L.J.; Arnold, A.E. Fungal Endophytes of Aquatic Macrophytes: Diverse host-generalists characterized by tissue preferences and geographic structure. Microb. Ecol. 2014, 67, 735–747. [Google Scholar] [CrossRef]
- He, D.; Shen, W.; Eberwein, J.; Zhao, Q.; Ren, L.; Wu, Q.L. Diversity and co-occurrence network of soil fungi are more responsive than those of bacteria to shifts in precipitation seasonality in a subtropical forest. Soil Biol. Biochem. 2017, 115, 499–510. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef]
- Toju, H.; Kishida, O.; Katayama, N.; Takagi, K. Networks depicting the fine-scale co-occurrences of fungi in soil horizons. PLoS ONE 2016, 11, e0165987. [Google Scholar] [CrossRef] [Green Version]
- De Menezes, A.B.; Richardson, A.E.; Thrall, P.H. Linking fungal–bacterial co-occurrences to soil ecosystem function. Curr. Opin. Microbiol. 2017, 37, 135–141. [Google Scholar] [CrossRef]
- Xue, L.; Ren, H.; Brodribb, T.J.; Wang, J.; Yao, X.; Li, S. Long term effects of management practice intensification on soil microbial community structure and co-occurrence network in a non-timber plantation. Forest Ecol. Manag. 2020, 459, 117805. [Google Scholar] [CrossRef]
- Lu, L.; Yin, S.; Liu, X.; Zhang, W.; Gu, T.; Shen, Q.; Qiu, H.Z. Fungal networks in yield-invigorating and -debilitating soils induced by prolonged potato monoculture. Soil Biol. Biochem. 2013, 65, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Karimi, B.; Maron, P.A.; Chemidlin-Prevost, B.N.; Bernard, N.; Gilbert, D.; Ranjard, L. Microbial diversity and ecological networks as indicators of environmental quality. Environ. Chem. Lett. 2017, 15, 265–281. [Google Scholar] [CrossRef]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence elationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Thrall, P.H.; Bissett, A.; van der Heijden, M.G.A.; Richardson, A.E. Linking microbial co-occurrences to soil ecological processes across a woodland-grassland ecotone. Ecol. Evol. 2018, 8, 8217–8230. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Qiang, W.; Liu, H.Y.; Ge, J.L.; Zuo, Y.L.; He, X.L. Effects of plants pecies on diversity of arbuscular mycorrhizal fungi in extremely arid desert environment. Mycosystema 2017, 36, 861–869. [Google Scholar] [CrossRef]
- Rodrigues, A.; Bacci, M.; Mueller, U.G.; Ortiz, A.; Pagnocca, F.C. Microfungal “weeds” in the leafcutter ant symbiosis. Microb. Ecol. 2008, 56, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Lugo, M.A.; Reinhart, K.O.; Menoyo, E.; Crespo, E.M.; Urcelay, C. Plant functional traits and phylogenetic relatedness explain variation in associations with root fungal endophytes in an extreme arid environment. Mycorrhiza 2014, 25, 85–95. [Google Scholar] [CrossRef]
- Xie, L.L.; He, X.L.; Wang, K.; Hou, L.F.; Sun, Q. Spatial dynamics of dark septate endophytes in the roots and rhizospheres of Hedysarum scoparium in northwest China and the influence of edaphic variables. Fungal Ecol. 2017, 26, 135–143. [Google Scholar] [CrossRef]
- Yu, J.; Xue, Z.K.; He, X.L.; Liu, C.M.; Steinberger, Y. Shifts in composition and diversity of arbuscular mycorrhizal fungi and glomalin contents during revegetation of desertified semiarid grassland. Appl. Soil Ecol. 2017, 115, 60–67. [Google Scholar] [CrossRef]
- Hou, L.F.; He, X.L.; Li, X.; Wang, S.J.; Zhao, L.L. Species composition and colonization of dark septate endophytes are affected by host plant species and soil depth in the Mu Us sandland, northwest China. Fungal Ecol. 2019, 39, 276–284. [Google Scholar] [CrossRef]
- Li, X.; He, C.; He, X.L.; Su, F.; Hou, L.F.; Ren, Y.; Hou, Y.T. Dark septate endophytes improve the growth of host and non-host plants under drought stress through altered root development. Plant Soil 2019, 439, 259–272. [Google Scholar] [CrossRef]
- Qiang, W.; He, X.L.; Wang, J.J.; Zhao, L.L. Temporal and spatial variation of arbuscular mycorrhizal fungi under the canopy of Hedysarum scoparium in the northern desert, China. Appl. Soil Ecol. 2019, 136, 139–147. [Google Scholar] [CrossRef]
- Kimmins, J.P. Forest Ecology; China Forestry Publishing House: Beijing, China, 2005; Volume 258, pp. 17–43. [Google Scholar]
- Yao, H.; Sun, X.; He, C.; Maitra, P.; Li, X.C.; Guo, L.D. Phyllosphere epiphytic and endophytic fungal community and network structures differ in a tropical mangrove ecosystem. Microbiome 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for Basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M., Gelfand, D., Sninsky, J., White, T., Eds.; Academic Press Inc.: New York, NY, USA, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Ihrmark, K.; Bödeker, I.T.M.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region-evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Dickie, I.A. Insidious effects of sequencing errors on perceived diversity in molecular surveys. New Phytol. 2010, 188, 916–918. [Google Scholar] [CrossRef]
- Dickie, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Jaramillo, J.E.; de Hollander, M.; Ramírez, C.A.; Mendes, R.; Raaijmakers, J.M.; Carrión, V.J. Deciphering rhizosphere microbiome assembly of wild and modern common bean (Phaseolus vulgaris) in native and agricultural soils from Colombia. Microbiome 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, K.R.; Somerfield, P.J.; Chapman, M.G. On resemblance measures for ecological studies, including taxonomic dissimilarities and a zero-adjusted Bray–Curtis coefficient for denuded assemblages. J. Exp. Mar. Biol. Ecol. 2006, 330, 55–80. [Google Scholar] [CrossRef]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B. Vegan: Community Ecology Package. R package Version 2.4. 2007; 4. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Toju, H.; Tanabe, A.S.; Ishii, H.S. Ericaceous plant-fungus network in a harsh alpine-subalpine environment. Mol. Ecol. 2016, 25, 3242–3257. [Google Scholar] [CrossRef]
- Toju, H.; Yamamoto, S.; Sato, H.; Tanabe, A.S. Sharing of diverse mycorrhizal and root-endophytic fungi among plant species in an oak dominated cool-temperate forest. PLoS ONE 2013, 8, e78248. [Google Scholar] [CrossRef]
- Blüthgen, N.; Menzel, F.; Hovestadt, T.; Fiala, B.; Blüthgen, N. Specialization, constraints, and conflicting interests in mutualistic networks. Curr. Biol. 2007, 17, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995, 85, 289–300. [Google Scholar] [CrossRef]
- Weiss, S.; Van Treuren, W.; Lozupone, C.; Faust, K.; Friedman, J.; Deng, Y.; Xia, L.C.; Xu, Z.Z.; Ursell, L.; Alm, E.J.; et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef]
- Chao, Y.; Liu, W.; Chen, Y.; Chen, W.; Zhao, L.; Ding, Q.; Wang, S.Z.; Tang, Y.T.; Zhang, T.; Qiu, R.L. Structure, variation, and co-occurrence of soil microbial communities in abandoned sites of a rare earth elements mine. Environ. Sci. Technol. 2016, 50, 11481–11490. [Google Scholar] [CrossRef]
- Banerjee, S.; Baah-Acheamfour, M.; Carlyle, C.N.; Bissett, A.; Richardson, A.E.; Siddique, T.; Bork, E.W.; Chang, S.X. Determinants of bacterial communities in Canadian agroforestry systems. Environ. Microbiol. 2016, 18, 1805–1816. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. Analyzing yeast protein–protein interaction data obtained from different sources. Nat. Biotechnol. 2002, 20, 991–997. [Google Scholar] [CrossRef]
- Tipton, L.; Müller, C.L.; Kurtz, Z.D.; Huang, L.; Kleerup, E.; Morris, A.; Bonneau, R.; Ghedin, E. Fungi stabilize connectivity in the lung and skin microbial ecosystems. Microbiome 2018, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.C.; Wang, J.H.; Zhao, M.; Liu, H.J.; Liao, K.T.; Xu, X.Y. Plant community and species diversity in the south fringe of Kumtag Desert. Chin. J. Plan. Ecol. 2006, 30, 375–382. [Google Scholar]
- Hardoim, P.R.; van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef] [Green Version]
- Porras-Alfaro, A.; Herrera, J.; Sinsabaugh, R.L.; Odenbach, K.J.; Lowrey, T.; Natvig, D.O. Novel root fungal consortium associated with a dominant desert grass. Appl. Environ. Microb. 2008, 74, 2805–2813. [Google Scholar] [CrossRef] [Green Version]
- González-Teuber, M.; Vilo, C.; Bascuñán-Godoy, L. Molecular characterization of endophytic fungi associated with the roots of Chenopodium quinoa inhabiting the Atacama Desert, Chile. Genom. Data 2017, 11, 109–112. [Google Scholar] [CrossRef]
- Hamm, P.; Mueller, R.C.; Kuske, C.R.; Porras-Alfaro, A. Keratinophilic fungi: Specialized fungal communities in a desert ecosystem identified using cultured-based and Illumina sequencing approaches. Microbiol. Res. 2020, 126530. [Google Scholar] [CrossRef]
- Canini, F.; Geml, J.; D’Acqui, L.P.; Buzzini, P.; Turchetti, B.; Onofri, S.; Ventura, S.; Zucconi, L. Fungal diversity and functionality are driven by soil texture in Taylor Valley, Antarctica. Fungal Ecol. 2021, 50, 101041. [Google Scholar] [CrossRef]
- De Menezes, G.C.A.; Câmara, P.E.A.S.; Pinto, O.H.B.; Carvalho-Silva, M.; Oliveira, F.S.; Souza, C.D.; Reynaud Schaefer, C.E.G.; Convey, P.; Rosa, C.A.; Rosa, L.H. Fungal diversity present on rocks from a polar desert in continental Antarctica assessed using DNA metabarcoding. Extremophiles 2021, 25, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, S.; Tojo, M.; Imura, S.; Herrero, M.L.; Uchida, M. Occurrence pattern of the parasitic fungus Rhytisma polare (Ascomycota) on the polar willow (Salix polaris) under limited water conditions in a high-Arctic semi-desert. Polar Biol. 2018, 41, 1105–1110. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Y.; Feng, W.; Qin, S.; Liu, Z. Revegetated shrub species recruit different soil fungal assemblages in a desert ecosystem. Plant Soil. 2019, 435, 81–93. [Google Scholar] [CrossRef]
- Wenndt, A.J.; Evans, S.E.; van Diepeningen, A.D.; Logan, J.R.; Jacobson, P.J.; Seely, M.K.; Jacobson, K.M. Why Plants Harbor Complex Endophytic Fungal Communities: Insights from Perennial Bunchgrass Stipagrostis sabulicola in the Namib Sand Sea. Front. Microbiol. 2021, 12, 691584. [Google Scholar] [CrossRef]
- Selbmann, L.; Egidi, E.; Isola, D.; Onofri, S.; Zucconi, L.; de Hoog, G.S.; Chinaglia, S.; Testa, L.; Tosi, S.; Balestrazzi, A.; et al. Biodiversity, evolution and adaptation of fungi in extreme environments. Plant Biosyst. 2013, 147, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Jia, T.; Wang, R.; Fan, X.; Chai, B. A comparative study of fungal community structure, diversity and richness between the soil and the phyllosphere of native grass species in a copper tailings dam in Shanxi Province, China. Appl. Sci. 2018, 8, 1297. [Google Scholar] [CrossRef] [Green Version]
- González-Menéndez, V.; Crespo, G.; de Pedro, N.; Diaz, C.; Martín, J.; Serrano, R.; Mackenzie, T.A.; Justicia, C.; González-Tejero, M.R.; Casares, M.; et al. Fungal endophytes from arid areas of Andalusia: High potential sources for antifungal and antitumoral agents. Sci. Rep. 2018, 8, 9729. [Google Scholar] [CrossRef] [Green Version]
- Loroa, M.; Valero-Jiménez, C.A.; Nozawa, S.; Márquez, L.M. Diversity and composition of fungal endophytes in semiarid Northwest Venezuela. J. Arid Environ. 2012, 85, 46–55. [Google Scholar] [CrossRef]
- Zhang, Y.; Crous, P.W.; Schoch, C.L.; Hyde, K.D. Pleosporales. Fungal Divers. 2012, 53, 1–221. [Google Scholar] [CrossRef] [Green Version]
- Knapp, D.G.; Kovács, G.M.; Zajta, E.; Groenewald, J.Z.; Crous, P.W. Dark septate endophytic pleosporalean genera from semiarid areas. Persoonia 2015, 35, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Aletaha, R.; Safari, S.A.A.; Zafari, D. A survey on endophytic fungi within roots of Chenopodiaceae species under different environmental conditions. Mycosphere 2018, 9, 618–634. [Google Scholar] [CrossRef]
- Bondarenko, S.A.; Georgieva, M.L.; Bilanenko, E.N. Fungi Inhabiting the Coastal Zone of Lake Magadi. Contemp. Probl. Ecol. 2018, 11, 439–448. [Google Scholar] [CrossRef]
- Korolyova, O.V. Species composition of Dothideomycetes in the anthropogenically transformed ecosystems of the steppe zone of Ukraine. Biosyst. Divers. 2017, 25, 4. [Google Scholar] [CrossRef]
- Hubert, N.A.; Gehring, C.A. Neighboring trees affect ectomycorrhizal fungal community composition in a woodland-forest ecotone. Mycorrhiza 2008, 18, 363–374. [Google Scholar] [CrossRef]
- Ghobad-Nejhad, M.; Dai, Y.C. Diplomitoporus rimosus is found in Asia and belongs to the Hymenochaetales. Mycologia 2010, 102, 1510–1517. [Google Scholar] [CrossRef]
- Richardson, S.N.; Walker, A.K.; Nsiama, T.K.; McFarlane, J.; Sumarah, M.W.; Ibrahim, A.; Miller, J.D. Griseofulvin-producing Xylaria endophytes of Pinus strobus and Vaccinium angustifolium: Evidence for a conifer-understory species endophyte ecology. Fungal Ecol. 2014, 11, 107–113. [Google Scholar] [CrossRef]
- Kemler, M.; Wingfield, M.J.; Cowan, D.A.; Slippers, B. Foliar fungi of the enigmatic desert plant Welwitschia mirabilis show little adaptation to their unique host plant. S. Afr. J. Sci. 2021, 117. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Wisniewski, M.; Schena, L.; Tack, A.J.M. Experimental evidence of microbial inheritance in plants and transmission routes from seed to phyllosphere and root. Environ. Microbiol. 2021. [Google Scholar] [CrossRef]
- Foster, K.R.; Schluter, J.; Coyte, K.Z.; Rakoff-Nahoum, S. The evolution of the host microbiome as an ecosystem on a leash. Nature 2017, 548, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genom. Biol. 2013, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Larkin, B.G.; Hunt, L.S.; Ramsey, P.W. Foliar nutrients shape fungal endophyte communities in Western white pine (Pinus monticola) with implications for white-tailed deer herbivory. Fungal Ecol. 2012, 5, 252–260. [Google Scholar] [CrossRef]
- Vincent, J.B.; Weiblen, G.D.; May, G. Host associations and beta diversity of fungal endophyte communities in New Guinea rainforest trees. Mol. Ecol. 2016, 25, 825–841. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [Green Version]
- Lau, M.K.; Arnold, A.E.; Johnson, N.C. Factors influencing communities of fungal endophytes in riparian woody plants. Fungal Ecol. 2013, 6, 365–378. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Spiering, M.J.; Scott, V.; Lane, G.A.; Christensen, M.J.; Schmid, J. In planta regulation of extension of an endophytic fungus and maintenance of high metabolic rates in its mycelium in the absence of apical extension. Appl. Environ. Microb. 2001, 67, 5377–5383. [Google Scholar] [CrossRef] [Green Version]
- Schardl, C.L.; Leuchtmann, A.; Spiering, M.J. Symbioses of grasses with seedborne fungal endophytes. Annu. Rev. Plant Boil. 2004, 55, 315–340. [Google Scholar] [CrossRef]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The soil microbiome influences grapevine-associated microbiota. MBio 2015, 6, e02527-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Glavina del Rio, T.; et al. Defning the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Wearn, J.A.; Sutton, B.C.; Morley, N.J.; Gange, A.C. Species and organ specificity of fungal endophytes in herbaceous grassland plants. J. Ecol. 2012, 100, 1085–1092. [Google Scholar] [CrossRef]
- Wu, L.; Han, T.; Li, W.; Jia, M.; Xue, L.; Rahman, K.; Qin, L.P. Geographic and tissue influences on endophytic fungal communities of Taxus chinensis var. mairei in China. Curr. Microbiol. 2013, 66, 40–48. [Google Scholar] [CrossRef]
- Petrini, O.; Sieber, T.N.; Toti, L.; Viret, O. Ecology, metabolite production, and substrate utilization in endophytic fungi. Nat. Toxins 1993, 1, 185–196. [Google Scholar] [CrossRef]
- Knapp, D.G.; Kovács, G.M. Interspecific metabolic diversity of root-colonizing endophytic fungi revealed by enzyme activity tests. FEMS Microbiol. Ecol. 2016, 92, fiw190. [Google Scholar] [CrossRef] [Green Version]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef]
- Mandakovic, D.; Rojas, C.; Maldonado, J.; Latorre, M.; Travisany, D.; Delage, E.; Bihouée, A.; Jean, G.; Díaz, F.P.; Fernández-Gómez, B.; et al. Structure and co-occurrence patterns in microbial communities under acute environmental stress reveal ecological factors fostering resilience. Sci. Rep. 2018, 8, 5875. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, F.; Marasco, R.; Fusi, M.; Scaglia, B.; Tsiamis, G.; Rolli, E.; Fodelianakis, S.; Bourtzis, K.; Ventura, S.; Tambone, F.; et al. The stage of soil development modulates rhizosphere effect along a High Arctic desert chronosequence. ISME J. 2018, 12, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Bissett, A.; Brown, M.V.; Siciliano, S.D.; Thrall, P.H. Microbial community responses to anthropogenically induced environmental change: Towards a systems approach. Ecol. Lett. 2013, 16, 128–139. [Google Scholar] [CrossRef]
- Eldridge, D.J.; Woodhouse, J.N.; Curlevski, N.J.A.; Hayward, M.; Brown, M.V.; Neilan, B.A. Soil-foraging animals alter the composition and co-occurrence of microbial communities in a desert shrubland. ISME J. 2015, 9, 2671–2681. [Google Scholar] [CrossRef] [Green Version]
- Větrovský, T.; Morais, D.; Kohout, P.; Lepinay, C.; Algora, C.; Hollá, S.A.; Bahnmann, B.D.; Bílohnědá, K.; Brabcová, V.; D’Alò, F.; et al. GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies. Sci. Data 2020, 7, 228. [Google Scholar] [CrossRef] [PubMed]
- Gond, S.K.; Mishra, A.; Sharma, V.K.; Verma, S.K.; Kumar, J.; Kharwar, R.N.; Kumar, A. Diversity and antimicrobial activity of endophytic fungi isolated from Nyctanthes arbortristis, a well-known medicinal plant of India. Mycoscience 2012, 53, 113–121. [Google Scholar] [CrossRef]
- Chen, L.; Saixi, Y.; Yi, R.; Baoyin, T. Characterization of soil microbes associated with a grazing-tolerant grass species, Stipa breviflora, in the Inner Mongolian desert steppe. Ecol. Evol. 2020, 10, 10607–10618. [Google Scholar] [CrossRef] [PubMed]
- Batta, Y. Entomopathogenic effect of Trichothecium roseum (Pers.) link (Hypocreales: Ascomycota) against Pauropsylla buxtoni (Psylloidea: Hemiptera) infesting Ficus carica leaves and its potential use as biocontrol agent of the insect. J. Appl. Microbiol. 2020, 129, 400–410. [Google Scholar] [CrossRef]
- Resquín-Romero, G.; Cabral-Antúnez, C.; Sarubbi-Orue, H.; Garrido-Jurado, I.; Valverde-García, P.; Schade, M.; Butt, T.M. Virulence of Metarhizium brunneum (Ascomycota: Hypocreales) strains against stinkbugs Euschistus heros and Dichelops furcatus (Hemiptera: Pentatomidae). J. Econ. Entomol. 2020, 113, 2540–2545. [Google Scholar] [CrossRef]
- White, R.L.; Geden, C.J.; Kaufman, P.E. Exposure timing and method affect Beauveria bassiana (Hypocreales: Cordycipitaceae) efficacy against house fly (Diptera: Muscidae) Larvae. J. Med. Entomol. 2020, 58, 372–378. [Google Scholar] [CrossRef]
- Harris, R. Effect of water potential on microbial growth and activity. In Water Potential Relations in Soil Microbiology; Parr, J., Gardner, W., Elliott, L., Eds.; Soil Science Society of America: Madison, WI, USA, 1981; pp. 23–95. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Species | Compartment Niches | |||
|---|---|---|---|---|
| Niche | R2 (%) | Pr (>F) | R2 (%) | Pr (>F) |
| Stem | 51.4 | 0.001 | 10.6 | 0.001 |
| Leaf | 41.1 | 0.004 | ||
| Root | 38.7 | 0.014 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, Y.; Li, X.; Yang, J.; Liu, J.; Zhao, L.; He, X. Fungal Endophytic Community and Diversity Associated with Desert Shrubs Driven by Plant Identity and Organ Differentiation in Extremely Arid Desert Ecosystem. J. Fungi 2021, 7, 578. https://doi.org/10.3390/jof7070578
Zuo Y, Li X, Yang J, Liu J, Zhao L, He X. Fungal Endophytic Community and Diversity Associated with Desert Shrubs Driven by Plant Identity and Organ Differentiation in Extremely Arid Desert Ecosystem. Journal of Fungi. 2021; 7(7):578. https://doi.org/10.3390/jof7070578
Chicago/Turabian StyleZuo, Yiling, Xia Li, Jingya Yang, Jiaqiang Liu, Lili Zhao, and Xueli He. 2021. "Fungal Endophytic Community and Diversity Associated with Desert Shrubs Driven by Plant Identity and Organ Differentiation in Extremely Arid Desert Ecosystem" Journal of Fungi 7, no. 7: 578. https://doi.org/10.3390/jof7070578