
A Fungal Defensin Targets the SARS−CoV−2 Spike Receptor−Binding Domain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction and Analysis of the Complex between Micasin and RBD
2.2. Molecular Dynamics (MD) Simulations
2.3. Construction of Computational Model of (BH)−1
2.4. Oxidative Refolding of Chemically Synthesized Micasin Mutants
2.5. Renaturation of RBD from E. coli Inclusion Body
2.6. Circular Dichroism (CD) Spectroscopy Analysis
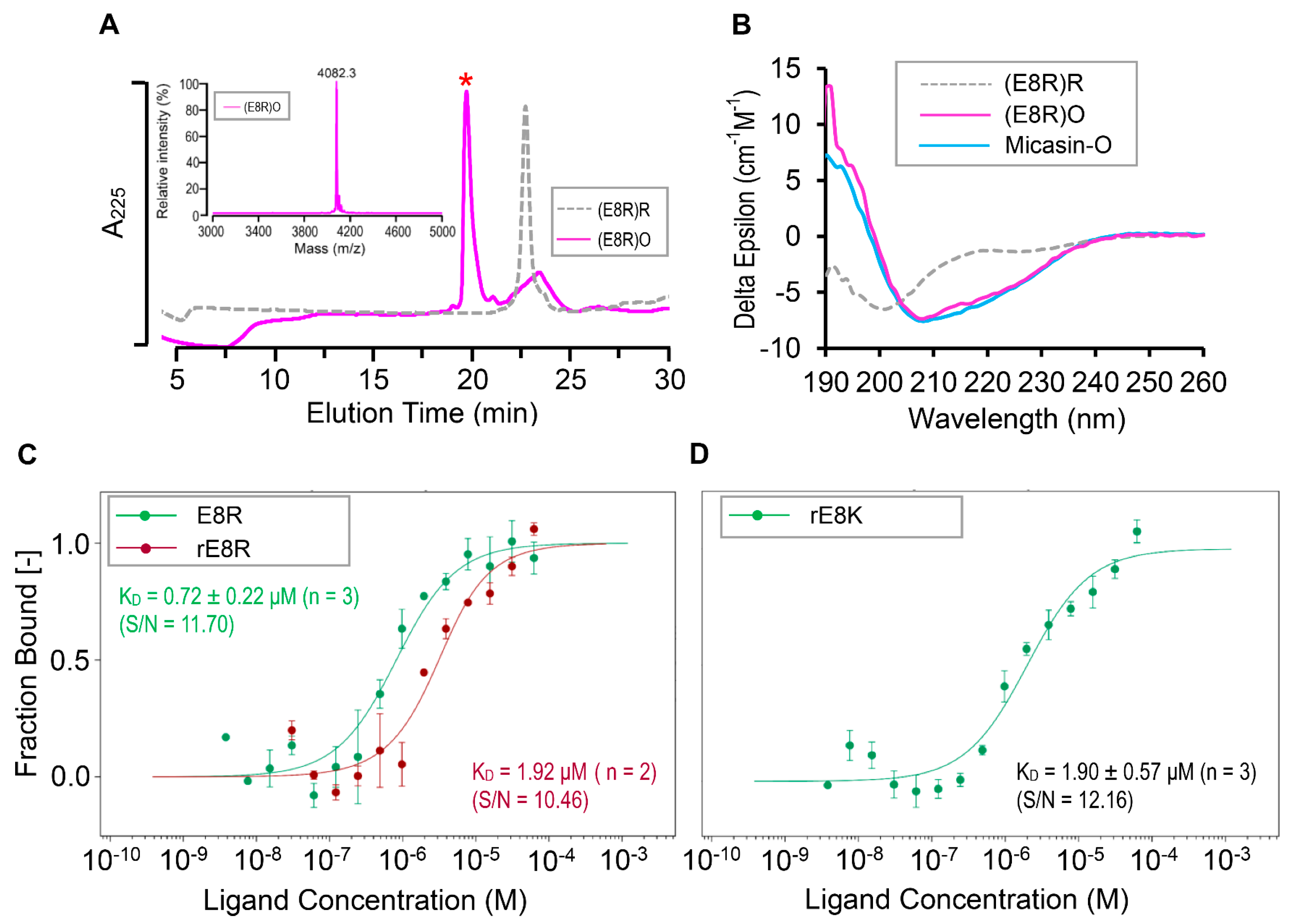
2.7. Determination of Peptide−RBD Binding Affinity Using MST
3. Results
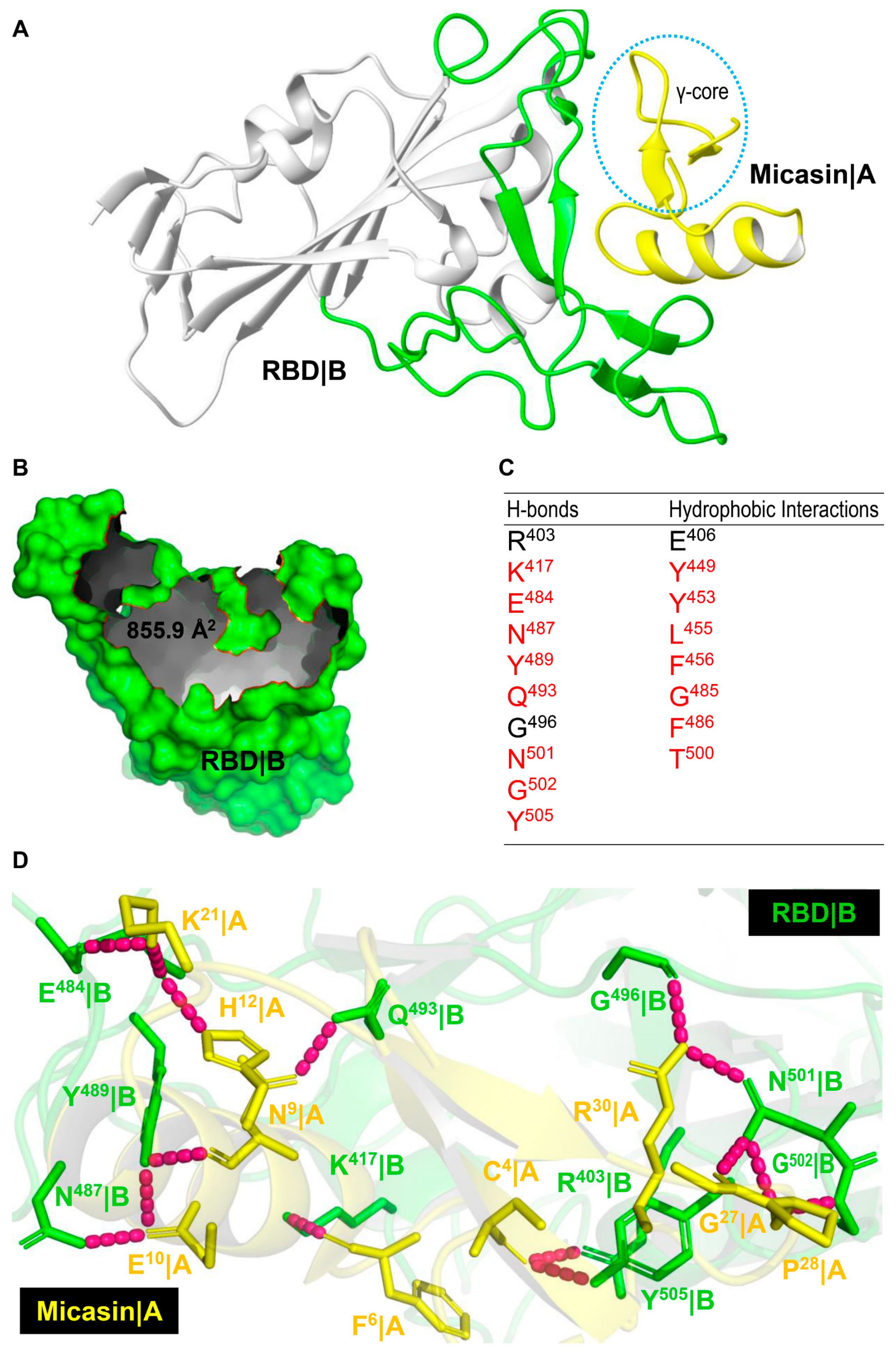
3.1. Computational Complex Revealing Extensive Shape Complementarity between Micasin and RBD
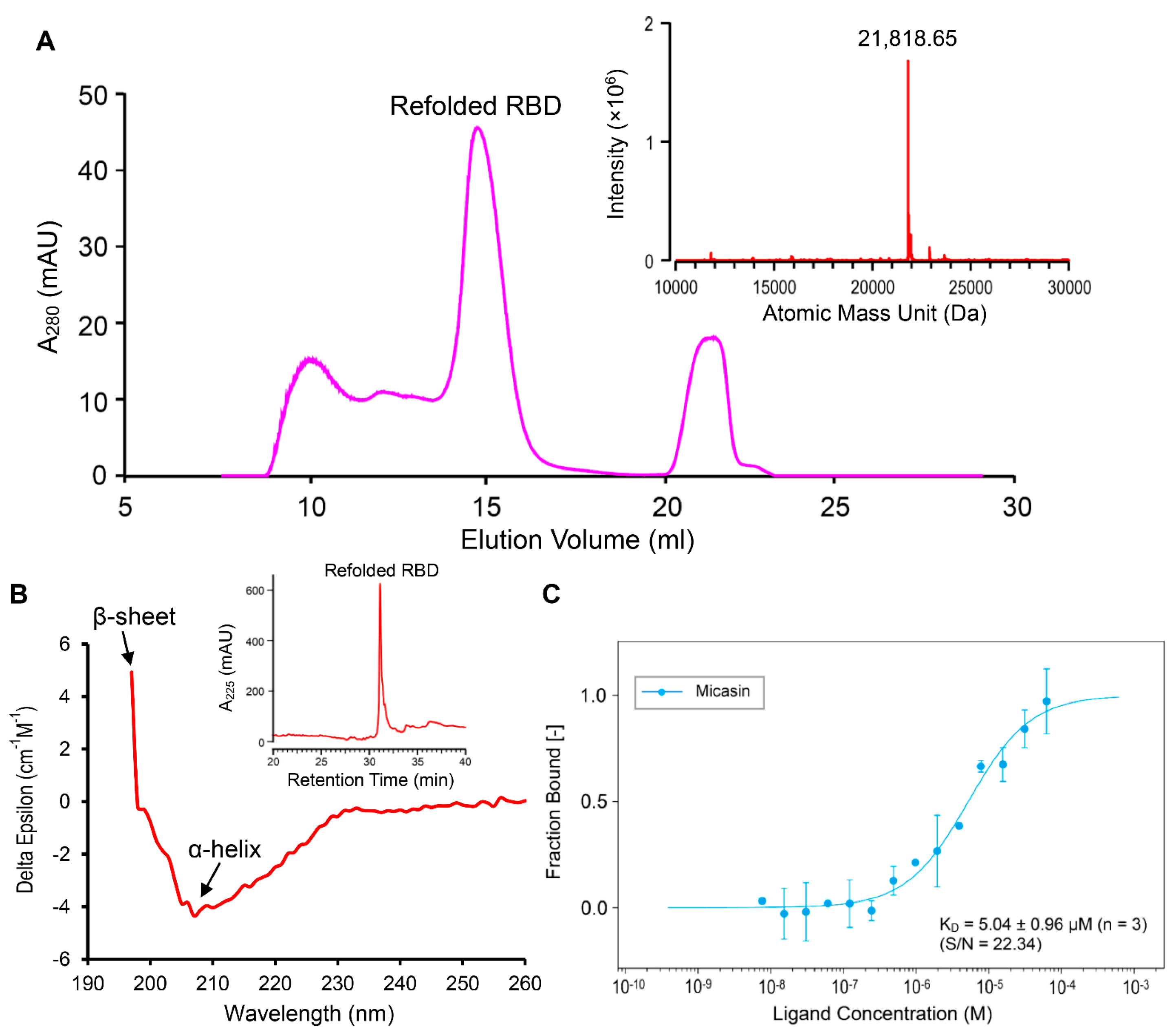
3.2. Micasin Binds RBD with Micromolar Affinity
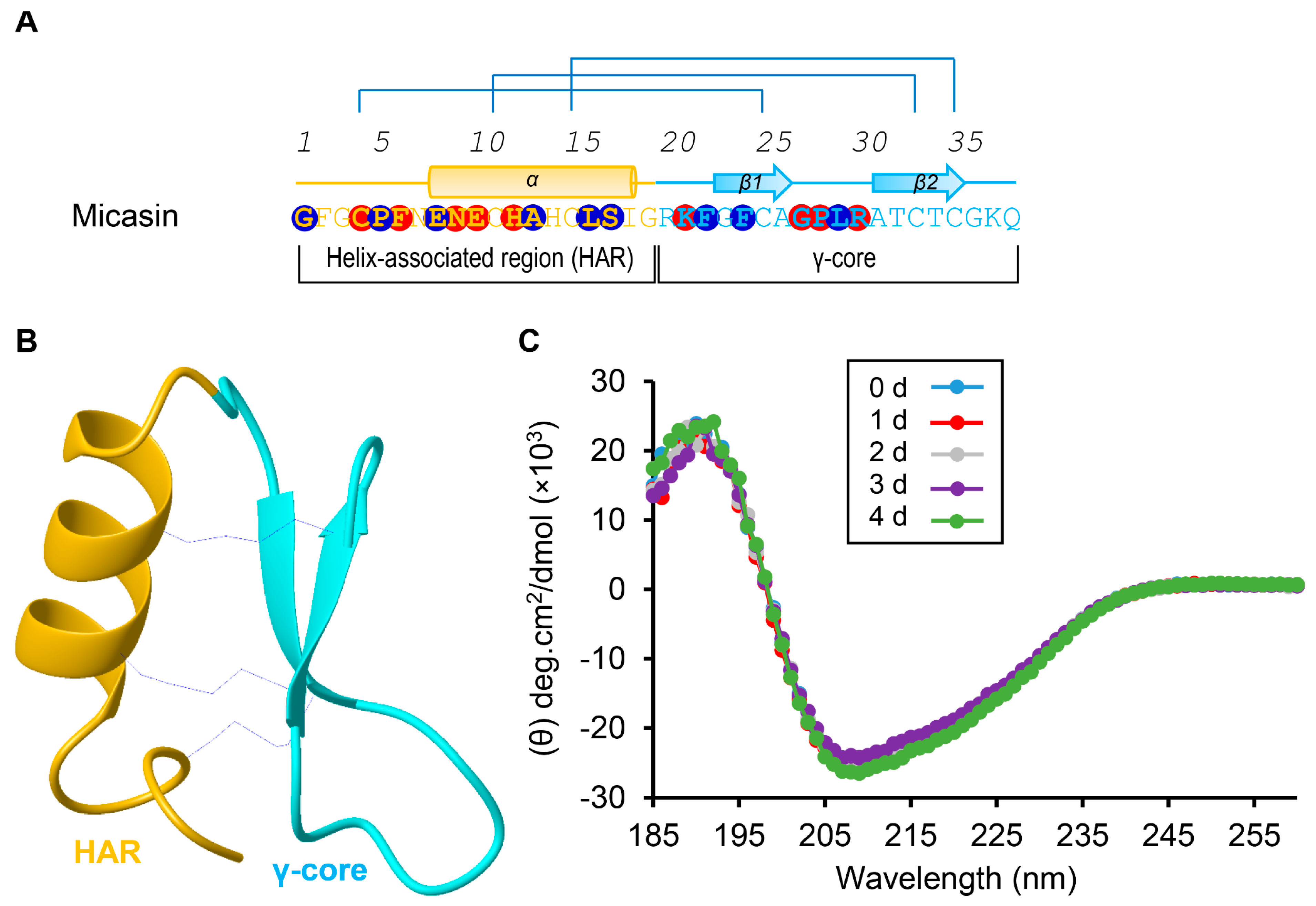
3.3. The Role of the γ−Core of Micasin in RBD Binding
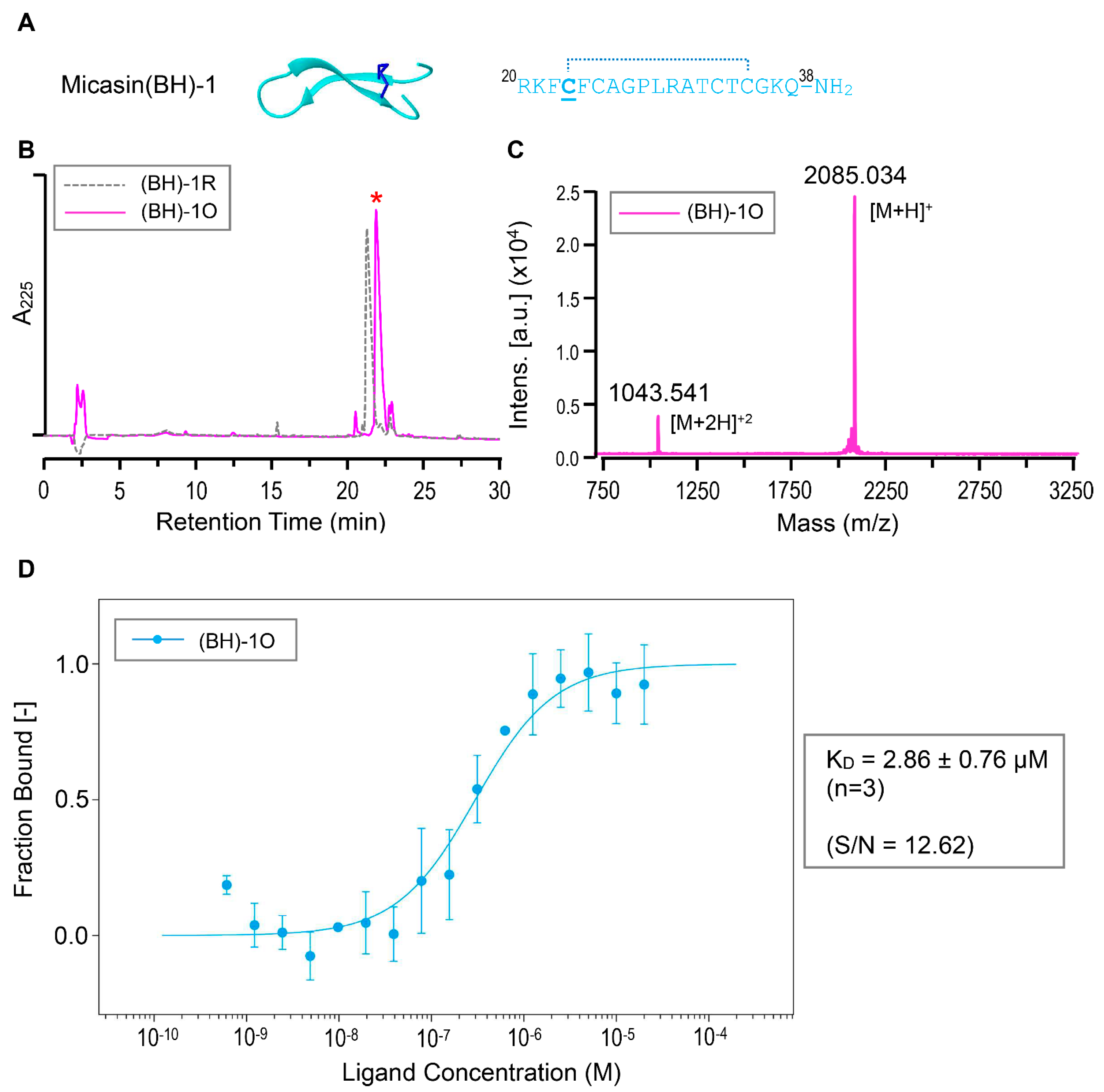
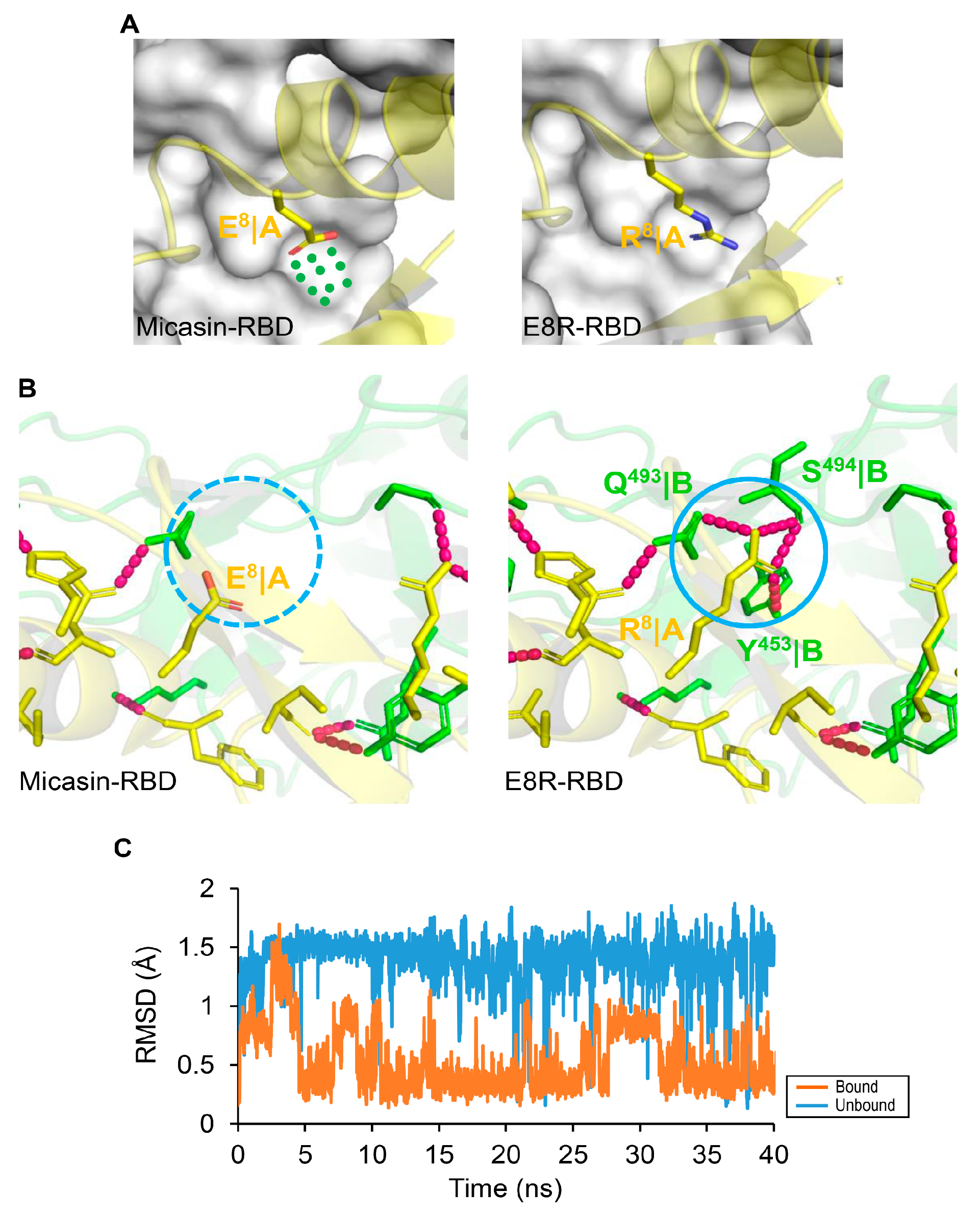
3.4. Structure−Based Design Improving the RBD Binding Affinity of Micasin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Gallagher, T. COVID19 therapeutics: Expanding the antiviral arsenal. EBioMedicine 2021, 66, 103289. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 Spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, B.J. A molecular trap against COVID-19. Science 2020, 369, 1167–1168. [Google Scholar] [CrossRef] [PubMed]
- Yavari, B.; Mahjub, R.; Saidijam, M.; Raigani, M.; Soleimani, M. The potential use of peptides in cancer treatment. Curr. Protein Pept. Sci. 2018, 19, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Curreli, F.; Victor, S.M.B.; Ahmed, S.; Drelich, A.; Tong, X.; Tseng, C.K.; Hillyer, C.D.; Debnath, A.K. Stapled peptides based on human angiotensin-converting enzyme 2 (ACE2) potently inhibit SARS-CoV-2 infection in vitro. mBio 2020, 11, e02451-20. [Google Scholar] [CrossRef]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Maiti, B.K. Potential role of peptide-based antiviral therapy against SARS-CoV-2 infection. ACS Pharmacol. Transl. Sci. 2020, 3, 783–785. [Google Scholar] [CrossRef]
- Kurpe, S.R.; Grishin, S.Y.; Surin, A.K.; Panfilov, A.V.; Slizen, M.V.; Chowdhury, S.D.; Galzitskaya, O.V. Antimicrobial and amyloidogenic activity of peptides. Can antimicrobial peptides be used against SARS-CoV-2? Int. J. Mol. Sci. 2020, 21, 9552. [Google Scholar] [CrossRef]
- Liscano, Y.; Oñate-Garzón, J.; Ocampo-Ibáñez, I.D. In silico discovery of antimicrobial peptides as an alternative to control SARS-CoV-2. Molecules 2020, 25, 5535. [Google Scholar] [CrossRef]
- Bakovic, A.; Risner, K.; Bhalla, N.; Alem, F.; Chang, T.L.; Weston, W.K.; Harness, J.A.; Narayanan, A. Brilacidin demonstrates inhibition of SARS-CoV-2 in cell culture. Viruses 2021, 13, 271. [Google Scholar] [CrossRef]
- Mousavi Maleki, M.S.; Restamian, M.; Madanchi, H. Antimicrobial peptides and other peptide-like therapeutics as promising candidates to combat SARS-CoV-2. Expert Rev. Anti Infect Ther. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Biragyn, A.; Hoover, D.M.; Lubkowski, J.; Oppenheim, J.J. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu. Rev. Immunol. 2004, 22, 181–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ghosh, S.K.; Basavarajappa, S.C.; Muller-Greven, J.; Penfield, J.; Brewer, A.; Ramakrishnan, P.; Buck, M.; Weinberg, A. Molecular dynamics simulations and functional studies reveal that hBD-2 binds SARS-CoV-2 Spike RBD and blocks viral entry into ACE2 expressing cells. bioRxiv 2021, 425621. [Google Scholar] [CrossRef]
- Niv, Y. Defensin 5 for prevention of SARS-CoV-2 invasion and COVID-19 disease. Med. Hypotheses 2020, 143, 110244. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, S.; Li, D.; Wei, D.Q.; Zhao, J.; Wang, J. Human intestinal defensin 5 inhibits SARS-CoV-2 invasion by cloaking ACE2. Gastroenterology 2020, 159, 1145–1147. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, D.; Chen, P.; Han, S.; Zhao, G.; Chen, Y.; Zhao, J.; Xiong, J.; Qiu, J.; et al. Human cathelicidin inhibits SARS-CoV-2 infection: Killing two birds with one stone. ACS Infect. Dis. 2021, 7, 1545–1554. [Google Scholar] [CrossRef]
- Zhao, H.; To, K.K.W.; Sze, K.H.; Yung, T.T.; Bian, M.; Lam, H.; Yeung, M.L.; Li, C.; Chu, H.; Yuen, K.Y. A broad-spectrum virus- and host-targeting peptide against respiratory viruses including influenza virus and SARS-CoV-2. Nat. Commun. 2020, 11, 4252. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gao, B.; Tytgat, J. Phylogenetic distribution, functional epitopes and evolution of the CSαβ superfamily. Cell. Mol. Life Sci. 2005, 62, 2257–2269. [Google Scholar] [CrossRef]
- Zhu, S. Discovery of six families of fungal defensin-like peptides provides insights into origin and evolution of the CSαβ defensins. Mol. Immunol. 2008, 45, 828–838. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, B.; Harvey, P.J.; Craik, D.J. Dermatopytic defensin with antiinfective potential. Proc. Natl. Acad. Sci. USA 2012, 109, 8495–8500. [Google Scholar] [CrossRef] [Green Version]
- Shafee, T.M.; Lay, F.T.; Hulett, M.D.; Anderson, M.A. The defensins consist of two independent, convergent protein superfamilies. Mol. Biol. Evol. 2016, 33, 2345–2356. [Google Scholar] [CrossRef] [Green Version]
- Vriens, K.; Cammue, B.P.; Thevissen, K. Antifungal plant defensins: Mechanisms of action and production. Molecules 2014, 19, 12280–12303. [Google Scholar] [CrossRef] [Green Version]
- Oppedijk, S.F.; Martin, N.I.; Breukink, E. Hit em where it hurts: The growing and structurally diverse family of peptides that target lipid-II. Biochim. Biophys. Acta 2016, 1858, 947–957. [Google Scholar] [CrossRef]
- Ritchie, D.W. Evaluation of protein docking predictions using Hex 3.1 in CAPRI rounds 1 and 2. Proteins 2003, 52, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 29–32, 51–55. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, B.; Peigneur, S.; Tytgat, J. How a scorpion toxin selectively captures a prey sodium channel: The molecular and evolutionary basis uncovered. Mol. Biol. Evol. 2020, 37, 3149–3164. [Google Scholar] [CrossRef]
- Gao, B.; Zhu, S. Identification and characterization of the parasitic wasp Nasonia defensins: Positive selection targeting the functional region? Dev. Comp. Immunol. 2010, 34, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Zhu, S. An insect defensin-derived β-hairpin peptide with enhanced antibacterial activity. ACS Chem. Biol. 2014, 9, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Rooklin, D.; Modell, A.E.; Li, H.; Berdan, V.; Arora, P.S.; Zhang, Y. Targeting unoccupied surfaces on protein-protein interfaces. J. Am. Chem. Soc. 2017, 139, 15560–15563. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A.R. The hydrogen bond in molecular recognition. Trends Biochem. Sci. 1987, 12, 301–304. [Google Scholar] [CrossRef]
- Wu, J.; Gao, B.; Zhu, S. Single-point mutation-mediated local amphipathic adjustment dramatically enhances antibacterial activity of a fungal defensin. FASEB J. 2016, 30, 2602–2614. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Gao, B.; Zhu, S. The fungal defensin family enlarged. Pharmaceuticals 2014, 7, 866–880. [Google Scholar] [CrossRef]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sönksen, C.P.; Ludvigsen, S.; Raventós, D.; Buskov, S.; Christensen, B.; De Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the Spike protein of SARS coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef]
- Lehmann, M.; Pasamontes, L.; Lassen, S.F.; Wyss, M. The consensus concept for thermostability engineering of proteins. Biochim. Biophys. Acta. 2000, 1543, 408–415. [Google Scholar] [CrossRef]
- Fowler, D.M.; Fields, S. Deep mutational scanning: A new style of protein science. Nat. Methods 2014, 11, 801–807. [Google Scholar] [CrossRef]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as protein engineering tool: Better than random based approaches? Comput. Struct. Biotechnol. 2018, 16, 25–33. [Google Scholar] [CrossRef]
- Gumulya, Y.; Gillam, E.M. Exploring the past and the future of protein evolution with ancestral sequence reconstruction: The ‘retro’ approach to protein engineering. Biochem. J. 2017, 474, 1–19. [Google Scholar] [CrossRef]
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 28046–28055. [Google Scholar] [CrossRef]
- Pack, P.; Plückthun, A. Miniantibodies: Use of amphipathic helices to produce functional, flexibly linked dimeric FV fragments with high avidity in Escherichia coli. Biochemistry 1992, 31, 1579–1584. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-based medicines: A review of fda-approved materials and clinical trials to date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.; Savani, R.C.; Zaki, H. SARS-CoV-2 Spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. bioRxiv 2021, 17, 435700. [Google Scholar]
- Colunga Biancatelli, R.; Solopov, P.; Sharlow, E.R.; Lazo, J.S.; Marik, P.E.; Catravas, J.D. The SARS-CoV-2 Spike Protein Subunit 1 induces COVID-19-like acute lung injury in Κ18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am. J. Physiol Lung Cell Mol. Physiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Asandei, A.; Mereuta, L.; Schiopu, I.; Park, J.; Seo, C.H.; Park, Y.; Luchian, T. Non-receptor-mediated lipid membrane permeabilization by the SARS-CoV-2 Spike protein S1 subunit. ACS Appl Mater. Interfaces. 2020, 12, 55649–55658. [Google Scholar] [CrossRef] [PubMed]
- Kubánková, M.; Hohberger, B.; Hoffmanns, J.; Fürst, J.; Herrmann, M.; Guck, J.; Kräter, M. Physical phenotype of blood cells is altered in COVID-19. Biophys. J. 2021. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, B.; Zhu, S. A Fungal Defensin Targets the SARS−CoV−2 Spike Receptor−Binding Domain. J. Fungi 2021, 7, 553. https://doi.org/10.3390/jof7070553
Gao B, Zhu S. A Fungal Defensin Targets the SARS−CoV−2 Spike Receptor−Binding Domain. Journal of Fungi. 2021; 7(7):553. https://doi.org/10.3390/jof7070553
Chicago/Turabian StyleGao, Bin, and Shunyi Zhu. 2021. "A Fungal Defensin Targets the SARS−CoV−2 Spike Receptor−Binding Domain" Journal of Fungi 7, no. 7: 553. https://doi.org/10.3390/jof7070553