
Structural Insight into a Yeast Maltase—The BaAG2 from Blastobotrys adeninivorans with Transglycosylating Activity

 , ,
, ,  , and
, and 
Abstract
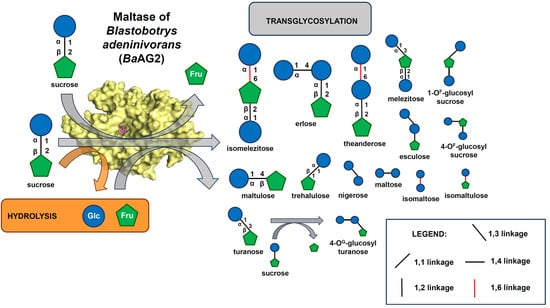
:
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Crystallization of the Maltase
2.3. Data Collection and Structure Determination
2.4. Transglycosylation Reaction and Product Separation
2.5. Nuclear Magnetic Resonance (NMR)
2.6. Thin Layer Chromatography (TLC) and High Performance Liquid Chromatography (HPLC)
2.7. Alignment of Protein Sequences
3. Results
3.1. In Silico Analysis of Maltase Sequences from Blastobotrys Species
3.2. Overall Quality and General Characterization of the BaAG2 Maltase Structure
3.3. Characterization of the Transglycosylation Activity of BaAG2 on Sucrose
3.4. Identification and Structural Determination of the Transglycosylation Products of Sucrose
4. Discussion
4.1. The Architecture of the Substrate-Binding Pocket of BaAG2 Reveals the Binding Mode of the Ligand and Exhibits Water Channels
4.2. The Active Center of the BaAG2 Is Similar to That of the Bacterial Maltase, but Differs from Enzymes Specialized to Hydrolyze Isomaltose-like Sugars
4.3. In Transglycosylation Reaction of BaAG2 on Sucrose a Variety of Bonds Are Synthesized
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Visnapuu, T.; Meldre, A.; Põšnograjeva, K.; Viigand, K.; Ernits, K.; Alamäe, T. Characterization of a Maltase from an Early-Diverged Non-Conventional Yeast Blastobotrys adeninivorans. Int. J. Mol. Sci. 2020, 21, 297. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Sanya, D.R.A.; Fouchard, F.; Nguyen, H.-V.; Kunze, G.; Neuvéglise, C.; Crutz-Le Coq, A.-M. Blastobotrys adeninivorans and B. raffinosifermentans, Two Sibling Yeast Species Which Accumulate Lipids at Elevated Temperatures and from Diverse Sugars. Biotechnol. Biofuels 2019, 12, 154. [Google Scholar] [CrossRef]
- Sanya, D.R.A.; Onésime, D.; Passoth, V.; Maiti, M.K.; Chattopadhyay, A.; Khot, M.B. Yeasts of the Blastobotrys Genus Are Promising Platform for Lipid-Based Fuels and Oleochemicals Production. Appl. Microbiol. Biotechnol. 2021, 105, 4879–4897. [Google Scholar] [CrossRef] [PubMed]
- Kunze, G.; Gaillardin, C.; Czernicka, M.; Durrens, P.; Martin, T.; Böer, E.; Gabaldón, T.; Cruz, J.A.; Talla, E.; Marck, C.; et al. The Complete Genome of Blastobotrys (Arxula) adeninivorans LS3—A Yeast of Biotechnological Interest. Biotechnol. Biofuels 2014, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Malak, A.; Baronian, K.; Kunze, G. Blastobotrys (Arxula) adeninivorans: A Promising Alternative Yeast for Biotechnology and Basic Research. Yeast Chichester Engl. 2016, 33, 535–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartmann, T.; Krüger, A.; Adler, K.; Duc, B.M.; Kunze, I.; Kunze, G. Temperature-Dependent Dimorphism of the Yeast Arxula adeninivorans Ls3. Antonie Van Leeuwenhoek 1995, 68, 215–223. [Google Scholar] [CrossRef]
- Bischoff, F.; Litwińska, K.; Cordes, A.; Baronian, K.; Bode, R.; Schauer, F.; Kunze, G. Three New Cutinases from the Yeast Arxula adeninivorans That Are Suitable for Biotechnological Applications. Appl. Environ. Microbiol. 2015, 81, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Böer, E.; Bode, R.; Mock, H.-P.; Piontek, M.; Kunze, G. Atan1p-an Extracellular Tannase from the Dimorphic Yeast Arxula adeninivorans: Molecular Cloning of the ATAN1 Gene and Characterization of the Recombinant Enzyme. Yeast Chichester Engl. 2009, 26, 323–337. [Google Scholar] [CrossRef]
- Bui, D.M.; Kunze, I.; Horstmann, C.; Schmidt, T.; Breunig, K.D.; Kunze, G. Expression of the Arxula adeninivorans Glucoamylase Gene in Kluyveromyces lactis. Appl. Microbiol. Biotechnol. 1996, 45, 102–106. [Google Scholar] [CrossRef]
- Böer, E.; Wartmann, T.; Luther, B.; Manteuffel, R.; Bode, R.; Gellissen, G.; Kunze, G. Characterization of the AINV Gene and the Encoded Invertase from the Dimorphic Yeast Arxula adeninivorans. Antonie Van Leeuwenhoek 2004, 86, 121–134. [Google Scholar] [CrossRef]
- Jankowska, D.A.; Trautwein-Schult, A.; Cordes, A.; Bode, R.; Baronian, K.; Kunze, G. A Novel Enzymatic Approach in the Production of Food with Low Purine Content Using Arxula adeninivorans Endogenous and Recombinant Purine Degradative Enzymes. Bioengineered 2015, 6, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Zeug, M.; Markovic, N.; Iancu, C.V.; Tripp, J.; Oreb, M.; Choe, J.-Y. Crystal Structures of Non-Oxidative Decarboxylases Reveal a New Mechanism of Action with a Catalytic Dyad and Structural Twists. Sci. Rep. 2021, 11, 3056. [Google Scholar] [CrossRef]
- Hasegawa, S.; Takizawa, M.; Suyama, H.; Shintani, T.; Gomi, K. Characterization and Expression Analysis of a Maltose-Utilizing (MAL) Cluster in Aspergillus oryzae. Fungal Genet. Biol. FG B 2010, 47, 1–9. [Google Scholar] [CrossRef]
- Fernández-Arrojo, L.; Marín, D.; Gómez De Segura, A.; Linde, D.; Alcalde, M.; Gutiérrez-Alonso, P.; Ghazi, I.; Plou, F.J.; Fernández-Lobato, M.; Ballesteros, A. Transformation of Maltose into Prebiotic Isomaltooligosaccharides by a Novel α-Glucosidase from Xantophyllomyces dendrorhous. Process Biochem. 2007, 42, 1530–1536. [Google Scholar] [CrossRef] [Green Version]
- Kato, N.; Suyama, S.; Shirokane, M.; Kato, M.; Kobayashi, T.; Tsukagoshi, N. Novel α-Glucosidase from Aspergillus nidulans with Strong Transglycosylation Activity. Appl. Environ. Microbiol. 2002, 68, 1250–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gonzalez, M.; Minguet-Lobato, M.; Plou, F.J.; Fernandez-Lobato, M. Molecular Characterization and Heterologous Expression of Two α-Glucosidases from Metschnikowia Spp, Both Producers of Honey Sugars. Microb. Cell Factories 2020, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, M.; Plou, F.J.; Cervantes, F.V.; Remacha, M.; Poveda, A.; Jiménez-Barbero, J.; Fernandez-Lobato, M. Efficient Production of Isomelezitose by a Glucosyltransferase Activity in Metschnikowia reukaufii Cell Extracts. Microb. Biotechnol. 2019, 12, 1274–1285. [Google Scholar] [CrossRef] [Green Version]
- Ouchemoukh, S.; Schweitzer, P.; Bachir Bey, M.; Djoudad-Kadji, H.; Louaileche, H. HPLC Sugar Profiles of Algerian Honeys. Food Chem. 2010, 121, 561–568. [Google Scholar] [CrossRef]
- Ruiz-Matute, A.I.; Brokl, M.; Soria, A.C.; Sanz, M.L.; Martínez-Castro, I. Gas Chromatographic–Mass Spectrometric Characterisation of Tri- and Tetrasaccharides in Honey. Food Chem. 2010, 120, 637–642. [Google Scholar] [CrossRef]
- Consonni, R.; Cagliani, L.R.; Cogliati, C. NMR Characterization of Saccharides in Italian Honeys of Different Floral Sources. J. Agric. Food Chem. 2012, 60, 4526–4534. [Google Scholar] [CrossRef]
- Chiba, S.; Murata, M.; Matsusaka, K.; Shimomura, T. A New Trisaccharide, 6F-α-d-Glucosyl-Sucrose, Synthesized by Transghicosylation Reaction of Brewer’s Yeast α-Glucosidase. Agric. Biol. Chem. 1979, 43, 775–779. [Google Scholar] [CrossRef]
- Henrissat, B.; Davies, G. Structural and Sequence-Based Classification of Glycoside Hydrolases. Curr. Opin. Struct. Biol. 1997, 7, 637–644. [Google Scholar] [CrossRef]
- Wierenga, R.K. The TIM-Barrel Fold: A Versatile Framework for Efficient Enzymes. FEBS Lett. 2001, 492, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal Structures of Isomaltase from Saccharomyces cerevisiae and in Complex with Its Competitive Inhibitor Maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, Y.; Mezaki, Y.; Kubota, M.; Matsuura, Y. Three-Dimensional Structure of Pseudomonas Isoamylase at 2.2 A Resolution. J. Mol. Biol. 1998, 281, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Steric Hindrance by 2 Amino Acid Residues Determines the Substrate Specificity of Isomaltase from Saccharomyces cerevisiae. J. Biosci. Bioeng. 2011, 112, 545–550. [Google Scholar] [CrossRef]
- Studier, F.W.; Moffatt, B.A. Use of Bacteriophage T7 RNA Polymerase to Direct Selective High-Level Expression of Cloned Genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Ursby, T.; Åhnberg, K.; Appio, R.; Aurelius, O.; Barczyk, A.; Bartalesi, A.; Bjelčić, M.; Bolmsten, F.; Cerenius, Y.; Doak, R.B.; et al. BioMAX—The First Macromolecular Crystallography Beamline at MAX IV Laboratory. J. Synchrotron Radiat. 2020, 27, 1415–1429. [Google Scholar] [CrossRef]
- Delagenière, S.; Brenchereau, P.; Launer, L.; Ashton, A.W.; Leal, R.; Veyrier, S.; Gabadinho, J.; Gordon, E.J.; Jones, S.D.; Levik, K.E.; et al. ISPyB: An Information Management System for Synchrotron Macromolecular Crystallography. Bioinforma. Oxf. Engl. 2011, 27, 3186–3192. [Google Scholar] [CrossRef] [Green Version]
- Incardona, M.-F.; Bourenkov, G.P.; Levik, K.; Pieritz, R.A.; Popov, A.N.; Svensson, O. EDNA: A Framework for Plugin-Based Applications Applied to X-Ray Experiment Online Data Analysis. J. Synchrotron Radiat. 2009, 16, 872–879. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbacher, R.; Godzik, A.; Grzechnik, S.K.; Jaroszewski, L. The Importance of Alignment Accuracy for Molecular Replacement. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1229–1236. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards Automated Crystallographic Structure Refinement with Phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. Publ. Protein Soc. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Schrödinger, LLC. Available online: https://www.schrodinger.com/products/pymol (accessed on 1 August 2021).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Mardo, K.; Visnapuu, T.; Vija, H.; Aasamets, A.; Viigand, K.; Alamäe, T. A Highly Active Endo-Levanase BT1760 of a Dominant Mammalian Gut Commensal Bacteroides thetaiotaomicron Cleaves Not Only Various Bacterial Levans, but Also Levan of Timothy Grass. PLoS ONE 2017, 12, e0169989. [Google Scholar] [CrossRef]
- Viigand, K.; Visnapuu, T.; Mardo, K.; Aasamets, A.; Alamäe, T. Maltase Protein of Ogataea (Hansenula) polymorpha Is a Counterpart to the Resurrected Ancestor Protein AncMALS of Yeast Maltases and Isomaltases. Yeast Chichester Engl. 2016, 33, 415–432. [Google Scholar] [CrossRef] [Green Version]
- Stingele, F.; Newell, J.W.; Neeser, J.R. Unraveling the Function of Glycosyltransferases in Streptococcus thermophilus Sfi6. J. Bacteriol. 1999, 181, 6354–6360. [Google Scholar] [CrossRef] [Green Version]
- Jork, H.; Funk, W.; Fischer, W.; Wimmer, H. Thin-Layer Chromatography. A: Physical and Chemical Detection Methods Fundamentals, Reagents I; VCH: Weinheim, Germany, 1990; Volume 1, ISBN 978-3-527-27834-3. [Google Scholar]
- Mardo, K.; Visnapuu, T.; Gromkova, M.; Aasamets, A.; Viigand, K.; Vija, H.; Alamäe, T. High-Throughput Assay of Levansucrase Variants in Search of Feasible Catalysts for the Synthesis of Fructooligosaccharides and Levan. Molecules 2014, 19, 8434–8455. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Shi, J.; Li, Y.; Gao, H.; Chang, L.; Zhang, Y.; Gao, L.; Xu, P.; Tang, S. Proteogenomics Study of Blastobotrys adeninivorans TMCC 70007-A Dominant Yeast in the Fermentation Process of Pu-Erh Tea. J. Proteome Res. 2021, 20, 3290–3304. [Google Scholar] [CrossRef]
- MacGregor, E.A.; Janecek, S.; Svensson, B. Relationship of Sequence and Structure to Specificity in the Alpha-Amylase Family of Enzymes. Biochim. Biophys. Acta 2001, 1546, 1–20. [Google Scholar] [CrossRef]
- Miyazaki, T.; Park, E.Y. Structure–Function Analysis of Silkworm Sucrose Hydrolase Uncovers the Mechanism of Substrate Specificity in GH13 Subfamily 17 Exo-α-Glucosidases. J. Biol. Chem. 2020, 295, 8784–8797. [Google Scholar] [CrossRef] [PubMed]
- Casa-Villegas, M.; Marín-Navarro, J.; Polaina, J. Amylases and Related Glycoside Hydrolases with Transglycosylation Activity Used for the Production of Isomaltooligosaccharides. Amylase 2018, 2, 17–29. [Google Scholar] [CrossRef]
- Auiewiriyanukul, W.; Saburi, W.; Kato, K.; Yao, M.; Mori, H. Function and Structure of GH13_31 α-Glucosidase with High α-(1→4)-Glucosidic Linkage Specificity and Transglucosylation Activity. FEBS Lett. 2018, 592, 2268–2281. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Saburi, W.; Gai, Z.; Kato, K.; Ojima-Kato, T.; Yu, J.; Komoda, K.; Kido, Y.; Matsui, H.; Mori, H.; et al. Structural Analysis of the α-Glucosidase HaG Provides New Insights into Substrate Specificity and Catalytic Mechanism. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Hondoh, H.; Saburi, W.; Mori, H.; Okuyama, M.; Nakada, T.; Matsuura, Y.; Kimura, A. Substrate Recognition Mechanism of Alpha-1,6-Glucosidic Linkage Hydrolyzing Enzyme, Dextran Glucosidase from Streptococcus mutans. J. Mol. Biol. 2008, 378, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, Y.; Kusunoki, M.; Harada, W.; Kakudo, M. Structure and Possible Catalytic Residues of Taka-Amylase A. J. Biochem. (Tokyo) 1984, 95, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Machius, M.; Wiegand, G.; Huber, R. Crystal Structure of Calcium-Depleted Bacillus licheniformis Alpha-Amylase at 2.2 A Resolution. J. Mol. Biol. 1995, 246, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Brayer, G.D.; Luo, Y.; Withers, S.G. The Structure of Human Pancreatic Alpha-Amylase at 1.8 A Resolution and Comparisons with Related Enzymes. Protein Sci. Publ. Protein Soc. 1995, 4, 1730–1742. [Google Scholar] [CrossRef] [PubMed]
- Harding, M.M. Small Revisions to Predicted Distances around Metal Sites in Proteins. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 678–682. [Google Scholar] [CrossRef]
- Giannesi, G.C.; de Lourdes Teixeira de Moraes Polizeli, M.; Terenzi, H.F.; Jorge, J.A. A Novel α-Glucosidase from Chaetomium thermophilum var. coprophilum That Converts Maltose into Trehalose: Purification and Partial Characterisation of the Enzyme. Process Biochem. 2006, 41, 1729–1735. [Google Scholar] [CrossRef]
- Kumara, H.M.; De Cort, S.; Verachtert, H. Localization and Characterization of Alpha-Glucosidase Activity in Brettanomyces lambicus. Appl. Environ. Microbiol. 1993, 59, 2352–2358. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.-M.; Liang, G.; Zhu, J.; Lu, B.; Peng, L.-X.; Wang, Q.-Y.; Wei, Y.-T.; Zhou, G.-P.; Huang, R.-B. Influence of Calcium Ions on the Thermal Characteristics of α-Amylase from Thermophilic Anoxybacillus Sp. GXS-BL. Protein Pept. Lett. 2019, 26, 148–157. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hondoh, H.; Mori, H.; Saburi, W.; Okuyama, M.; Kimura, A. Calcium Ion-Dependent Increase in Thermostability of Dextran Glucosidase from Streptococcus mutans. Biosci. Biotechnol. Biochem. 2011, 75, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Lee, S.B.; Lee, H.S.; Lee, S.Y.; Baek, J.S.; Kim, D.; Moon, T.W.; Robyt, J.F.; Park, K.H. Comparative Study of the Inhibition of Alpha-Glucosidase, Alpha-Amylase, and Cyclomaltodextrin Glucanosyltransferase by Acarbose, Isoacarbose, and Acarviosine-Glucose. Arch. Biochem. Biophys. 1999, 371, 277–283. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Davies, G.J. Structure of the Aspergillus oryzae Alpha-Amylase Complexed with the Inhibitor Acarbose at 2.0 A Resolution. Biochemistry 1997, 36, 10837–10845. [Google Scholar] [CrossRef] [PubMed]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human Intestinal Maltase-Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Strokopytov, B.; Penninga, D.; Rozeboom, H.J.; Kalk, K.H.; Dijkhuizen, L.; Dijkstra, B.W. X-Ray Structure of Cyclodextrin Glycosyltransferase Complexed with Acarbose. Implications for the Catalytic Mechanism of Glycosidases. Biochemistry 1995, 34, 2234–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrington, M.S.; Davis, S.N. Considerations When Using Alpha-Glucosidase Inhibitors in the Treatment of Type 2 Diabetes. Expert Opin. Pharmacother. 2019, 20, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Alonso, P.; Gimeno-Pérez, M.; Ramírez-Escudero, M.; Plou, F.J.; Sanz-Aparicio, J.; Fernández-Lobato, M. Molecular Characterization and Heterologous Expression of a Xanthophyllomyces dendrorhous α-Glucosidase with Potential for Prebiotics Production. Appl. Microbiol. Biotechnol. 2016, 100, 3125–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The Carbohydrate-Active Enzymes Database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Hata, Y.; Kizaki, H.; Katsube, Y.; Suzuki, Y. The Refined Crystal Structure of Bacillus cereus Oligo-1,6-Glucosidase at 2.0 A Resolution: Structural Characterization of Proline-Substitution Sites for Protein Thermostabilization. J. Mol. Biol. 1997, 269, 142–153. [Google Scholar] [CrossRef]
- Mirza, O.; Skov, L.K.; Remaud-Simeon, M.; Potocki de Montalk, G.; Albenne, C.; Monsan, P.; Gajhede, M. Crystal Structures of Amylosucrase from Neisseria polysaccharea in Complex with D-Glucose and the Active Site Mutant Glu328Gln in Complex with the Natural Substrate Sucrose. Biochemistry 2001, 40, 9032–9039. [Google Scholar] [CrossRef]
- Sprogøe, D.; van den Broek, L.A.M.; Mirza, O.; Kastrup, J.S.; Voragen, A.G.J.; Gajhede, M.; Skov, L.K. Crystal Structure of Sucrose Phosphorylase from Bifidobacterium adolescentis. Biochemistry 2004, 43, 1156–1162. [Google Scholar] [CrossRef]
- Kadziola, A.; Søgaard, M.; Svensson, B.; Haser, R. Molecular Structure of a Barley Alpha-Amylase-Inhibitor Complex: Implications for Starch Binding and Catalysis. J. Mol. Biol. 1998, 278, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Voordeckers, K.; Brown, C.A.; Vanneste, K.; van der Zande, E.; Voet, A.; Maere, S.; Verstrepen, K.J. Reconstruction of Ancestral Metabolic Enzymes Reveals Molecular Mechanisms Underlying Evolutionary Innovation through Gene Duplication. PLoS Biol. 2012, 10, e1001446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Nakayama, A.; Yamamoto, Y.; Tabata, S. Val216 Decides the Substrate Specificity of Alpha-Glucosidase in Saccharomyces cerevisiae. Eur. J. Biochem. 2004, 271, 3414–3420. [Google Scholar] [CrossRef] [PubMed]
- Viigand, K.; Põšnograjeva, K.; Visnapuu, T.; Alamäe, T. Genome Mining of Non-Conventional Yeasts: Search and Analysis of MAL Clusters and Proteins. Genes 2018, 9, 354. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Tanaka, H.; Takemura, R.; Yokogawa, T.; Shimonaka, A.; Matsui, H.; Kashiwabara, S.; Watanabe, K.; Suzuki, Y. Molecular Determinants of Substrate Recognition in Thermostable Alpha-Glucosidases Belonging to Glycoside Hydrolase Family 13. J. Biochem. (Tokyo) 2007, 142, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hasegawa, N.; Saito, J.; Umezawa, S.; Honda, Y.; Kino, K.; Kirimura, K. Purification, Characterization, and Gene Identification of an α-Glucosyl Transfer Enzyme, a Novel Type α-Glucosidase from Xanthomonas campestris WU-9701. J. Mol. Catal. B Enzym. 2012, 80, 20–27. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Saccharide | Composition and Linkage Type | 500 mM Sucrose | 1500 mM Sucrose |
|---|---|---|---|
| Sucrose—Substrate | Glc-α1,2-Fru | 43.2% | 69.4% |
| Maltulose | Glc-α1,4-Fru | 23.9% | 13.5% |
| Trehalulose | Glc-α1,1-Fru | 16.4% | 7.8% |
| Turanose | Glc-α1,3-Fru | 5.9% | 4.2% |
| Nigerose | Glc-α1,3-Glc | 3.7% | 1.9% |
| Maltose | Glc-α1,4-Glc | 3.0% | 1.0% |
| Isomaltose | Glc-α1,6-Glc | 2.1% | 1.7% |
| Isomaltulose (Palatinose) | Glc-α1,6-Fru | 1.8% | 0.4% |
| Saccharide | Composition and Linkage Type | 500 mM Sucrose | 1500 mM Sucrose |
|---|---|---|---|
| Isomelezitose * | Glc-α1,6-Fru-β2,1-Glc | 32.2% | 25.0% |
| Erlose | Glc-α1,4-Glc-α1,2-Fru | 25.8% | 31.7% |
| Theanderose * | Glc-α1,6-Glc-α1,2-Fru | 22.7% | 20.5% |
| Melezitose | Glc-α1,3-Fru-β2,1-Glc | 9.6% | 15.9% |
| 1-OF-Glucosyl-Sucrose | Glc-α1,1-Fru-β2,1-Glc | 3.7% | 2.7% |
| Esculose | Glc-α1,3-Glc-α1,2-Fru | 2.6% | 0.8% |
| 4-OG-Glucosyl-Turanose | Glc-α1,4-Glc-α1,3-Fru | 2.0% | 1.7% |
| 4-OF-Glucosyl-Sucrose | Glc-α1,4-Fru-β2,1-Glc | 1.4% | 1.7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ernits, K.; Kjeldsen, C.; Persson, K.; Grigor, E.; Alamäe, T.; Visnapuu, T. Structural Insight into a Yeast Maltase—The BaAG2 from Blastobotrys adeninivorans with Transglycosylating Activity. J. Fungi 2021, 7, 816. https://doi.org/10.3390/jof7100816
Ernits K, Kjeldsen C, Persson K, Grigor E, Alamäe T, Visnapuu T. Structural Insight into a Yeast Maltase—The BaAG2 from Blastobotrys adeninivorans with Transglycosylating Activity. Journal of Fungi. 2021; 7(10):816. https://doi.org/10.3390/jof7100816
Chicago/Turabian StyleErnits, Karin, Christian Kjeldsen, Karina Persson, Eliis Grigor, Tiina Alamäe, and Triinu Visnapuu. 2021. "Structural Insight into a Yeast Maltase—The BaAG2 from Blastobotrys adeninivorans with Transglycosylating Activity" Journal of Fungi 7, no. 10: 816. https://doi.org/10.3390/jof7100816