Congruence Amidst Discordance between Sequence and Protein-Content Based Phylogenies of Fungi


Abstract
:1. Introduction
2. Material and Methods
2.1. Examined Fungal Species
2.2. Pfam Analysis and Subcellular Location Prediction
2.3. Phylogenetic Tree Construction
2.4. Tree Topology Congruence Tests
3. Results and Discussion
3.1. Protein Family Size Variation
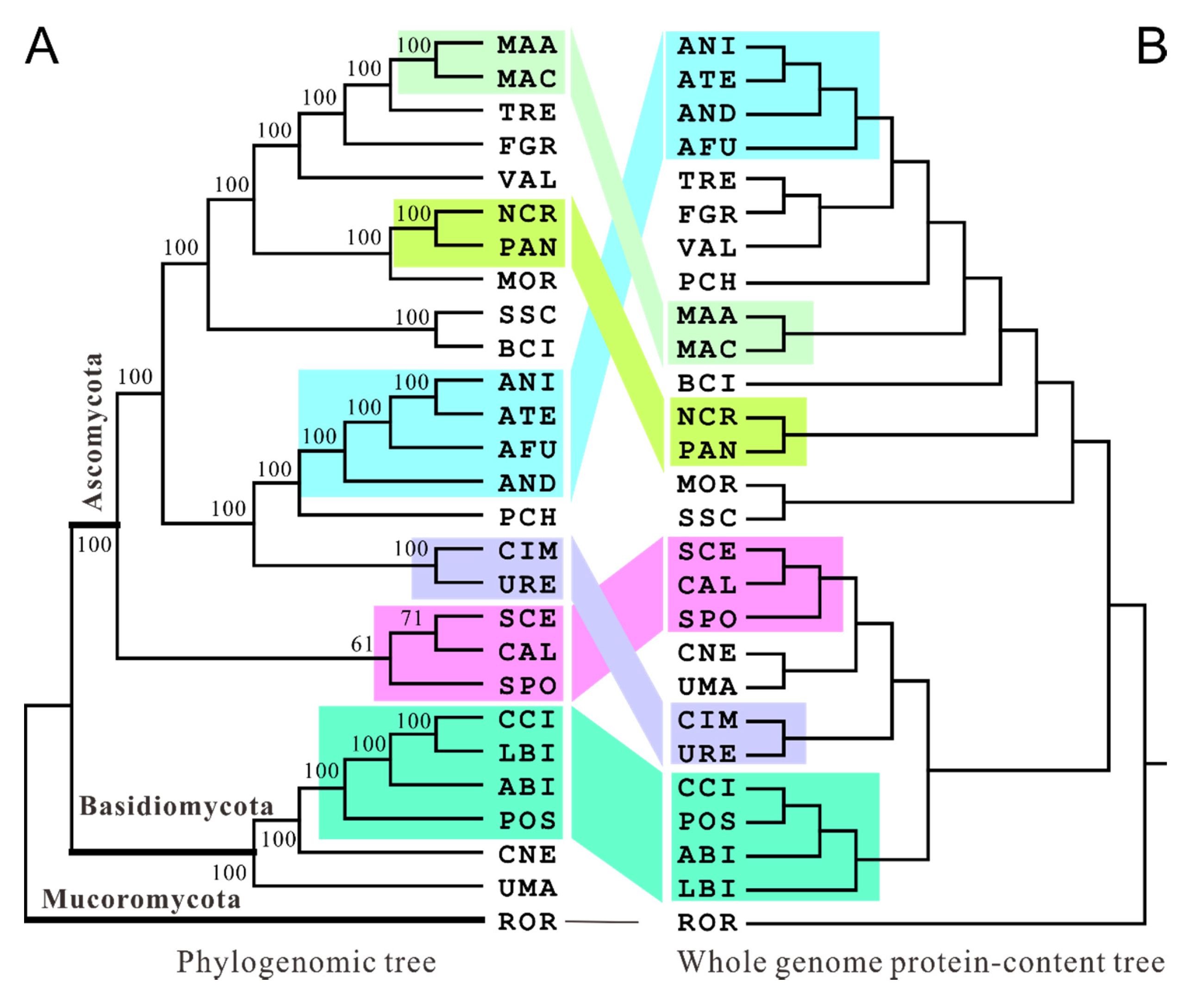
3.2. Diverse Phylogenetic Associations
3.3. Congruence and Discordance Relationships of Different Phylogenies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gabaldon, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorogood, C.J.; Bauer, U.; Hiscock, S.J. Convergent and divergent evolution in carnivorous pitcher plant traps. New Phytol. 2018, 217, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.F.; Xiao, G.H.; Zheng, P.; Cen, K.; Zhan, S.; Wang, C.S. Divergent and convergent evolution of fungal pathogenicity. Genome Biol. Evol. 2016, 8, 1374–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.; Kennedy, M.; Johnson, K.P.; Palma, R.L.; Page, R.D. Multiple cophylogenetic analyses reveal frequent cospeciation between pelecaniform birds and Pectinopygus lice. Syst. Biol. 2007, 56, 232–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Xiao, G.; Zheng, P.; Shang, Y.; Su, Y.; Zhang, X.; Liu, X.; Zhan, S.; St Leger, R.J.; Wang, C. Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc. Natl. Acad. Sci. USA 2014, 111, 16796–16801. [Google Scholar] [CrossRef] [Green Version]
- Snel, B.; Huynen, M.A.; Dutilh, B.E. Genome trees and the nature of genome evolution. Annu. Rev. Microbiol. 2005, 59, 191–209. [Google Scholar] [CrossRef] [Green Version]
- Pett, W.; Adamski, M.; Adamska, M.; Francis, W.R.; Eitel, M.; Pisani, D.; Wörheide, G. The role of homology and orthology in the phylogenomic analysis of Metazoan gene content. Mol. Biol. Evol. 2019, 36, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Snel, B.; Bork, P.; Huynen, M.A. Genome phylogeny based on gene content. Nat. Genet. 1999, 21, 108–110. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Oppenheim, S.; DeSalle, R. A whole genome gene content phylogenetic analysis of anopheline mosquitoes. Mol. Phylogenet. Evol. 2017, 107, 266–269. [Google Scholar] [CrossRef] [Green Version]
- Tekaia, F.; Lazcano, A.; Dujon, B. The genomic tree as revealed from whole proteome comparisons. Genome Res. 1999, 9, 550–557. [Google Scholar]
- Pisani, D.; Pett, W.; Dohrmann, M.; Feuda, R.; Rota-Stabelli, O.; Philippe, H.; Lartillot, N.; Wörheide, G. Genomic data do not support comb jellies as the sister group to all other animals. Proc. Natl. Acad. Sci. USA 2015, 112, 15402–15407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.F.; Pang, K.; Schnitzler, C.E.; Nguyen, A.D.; Moreland, R.T.; Simmons, D.K.; Koch, B.J.; Francis, W.R.; Havlak, P.; Smith, S.A.; et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 2013, 342, 1242592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montague, M.G.; Hutchison, C.A., 3rd. Gene content phylogeny of herpesviruses. Proc. Natl. Acad. Sci. USA 2000, 97, 5334–5339. [Google Scholar] [CrossRef] [Green Version]
- Fitz-Gibbon, S.T.; House, C.H. Whole genome-based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res. 1999, 27, 4218–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.W.; Brandt, B.W. ProFASTA: A pipeline web server for fungal protein scanning with integration of cell surface prediction software. Fungal Genet. Biol. 2012, 49, 173–179. [Google Scholar] [CrossRef]
- O’Brien, K.P.; Remm, M.; Sonnhammer, E.L. Inparanoid: A comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005, 33, D476–D480. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar]
- De Vienne, D.M.; Giraud, T.; Martin, O.C. A congruence index for testing topological similarity between trees. Bioinformatics 2007, 23, 3119–3124. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, R.J.; Sweeney, M.D.; Lo Leggio, L.; Otten, H.; Poulsen, J.C.; Johansen, K.S.; Krogh, K.B.; Jorgensen, C.I.; Tovborg, M.; Anthonsen, A.; et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. USA 2011, 108, 15079–15084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, S. Insect pathogenic fungi: Genomics, molecular interactions, and genetic improvements. Annu. Rev. Entomol. 2017, 62, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nadales, E.; Nogueira, M.F.; Baldin, C.; Castanheira, S.; El Ghalid, M.; Grund, E.; Lengeler, K.; Marchegiani, E.; Mehrotra, P.V.; Moretti, M.; et al. Fungal model systems and the elucidation of pathogenicity determinants. Fungal Genet. Biol. 2014, 70, 42–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.W.; Zhang, X.L.; Li, K.; Wang, C.S.; Cai, L.; Zhuang, W.Y.; Xiang, M.C.; Liu, X.Z. Introgression and gene family contraction drive the evolution of lifestyle and host shifts of hypocrealean fungi. Mycology 2018, 9, 176–188. [Google Scholar] [CrossRef]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martinez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.B.; St Leger, R.J.; Wang, C. Advances in Genomics of Entomopathogenic Fungi. Adv. Genet. 2016, 94, 67–105. [Google Scholar]
- Gibbons, J.G.; Rokas, A. The function and evolution of the Aspergillus genome. Trends Microbiol. 2013, 21, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet 2011, 7, e1002230. [Google Scholar] [CrossRef] [Green Version]
- Sharpton, T.J.; Stajich, J.E.; Rounsley, S.D.; Gardner, M.J.; Wortman, J.R.; Jordar, V.S.; Maiti, R.; Kodira, C.D.; Neafsey, D.E.; Zeng, Q.; et al. Comparative genomic analyses of the human fungal pathogens Coccidioides and their relatives. Genome Res. 2009, 19, 1722–1731. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, I.; Gtari, M.; Tisa, L.S.; Sen, A. A novel phylogenetic tree based on the presence of protein domains in selected actinobacteria. Antonie Leeuwenhoek 2019, 112, 101–107. [Google Scholar] [CrossRef]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog 2012, 8, e1003037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.C.; Chen, C.J.; Li, W.H.; Chuang, T.J. Gene family size conservation is a good indicator of evolutionary rates. Mol. Biol. Evol. 2010, 27, 1750–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronado, J.E.; Mneimneh, S.; Epstein, S.L.; Qiu, W.G.; Lipke, P.N. Conserved processes and lineage-specific proteins in fungal cell wall evolution. Eukaryot. Cell 2007, 6, 2269–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Jiang, H.; Zhao, X.; Lu, Z.; Luo, Z.; Li, X.; Zhao, J.; Zhang, Y. Correlation of cell surface proteins of distinct Beauveria bassiana cell types and adaption to varied environment and interaction with the host insect. Fungal Genet. Biol. 2017, 99, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.; Veses, V.; Gow, N.A.R. Vacuole dynamics in fungi. Fungal Biol. Rev. 2010, 24, 93–105. [Google Scholar] [CrossRef]
- Gu, X.; Zhang, H. Genome phylogenetic analysis based on extended gene contents. Mol. Biol. Evol. 2004, 21, 1401–1408. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.O. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 2001, 50, 913–925. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Sequence Tree | Genome-Wide Proteins | Proteases | CAZy ** | Ribosomal Subunits | |
|---|---|---|---|---|---|
| Whole genome proteins | 1.32; 0.0106 | ||||
| Proteases | 1.45; 1.36 × 10−3 | 1.19; 0.0835 | |||
| CAZy ** | 1.19; 0.0835 | 1.19; 0.0835 | 1.71; 2.20 × 10−5 | ||
| Ribosomal subunits | 1.05; 0.656 | 1.19; 0.0835 | 1.05; 0.656 | 1.19; 0.0835 | |
| DUFs and UPFs *** | 1.45; 1.36 × 10−3 | 1.71; 2.20 × 10−5 | 1.45; 1.36 × 10−3 | 2.11; 4.54 × 10−8 | 1.19; 0.0835 |
| Sequence Tree | WGP | CWP | NP | MP | CP | PMP | ERP | GP | EP | VP ** | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Whole genome proteins (WGP) | 1.32; 0.0106 | ||||||||||
| Cell wall protein (CWP) | 1.98; 3.56 × 10−7 | 1.32; 0.0106 | |||||||||
| Nuclear protein (NP) | 1.71; 2.20 × 10−5 | 1.58; 1.72 × 10−4 | 1.85; 2.80 × 10−6 | ||||||||
| Mitochondrial protein (MP) | 1.58; 1.72 × 10−4 | 1.85; 2.80 × 10−6 | 1.58; 1.72 × 10−4 | 1.85; 2.80 × 10−6 | |||||||
| Cytoplasmic protein (CP) | 1.45; 1.36 × 10−3 | 2.11; 4.54 × 10−8 | 1.06; 0.6560 | 1.32; 0.0106 | 1.71; 2.20 × 10−5 | ||||||
| Plasma membrane protein (PMP) | 1.45; 1.36 × 10−3 | 1.71; 2.20 × 10−5 | 1.85; 2.80 × 10−6 | 1.58; 1.72 × 10−4 | 1.98; 3.56 × 10−7 | 1.85; 2.80 × 10−6 | |||||
| Endoplasmic reticulum protein (ERP) | 1.32; 0.0106 | 1.45; 1.36 × 10−3 | 1.32; 0.0106 | 1.45; 1.36 × 10−3 | 1.98; 3.56 × 10−7 | 1.45; 1.36 × 10−3 | 1.85; 2.80 × 10−6 | ||||
| Golgi protein (GP) | 1.32; 0.0106 | 1.58; 1.73 × 10−4 | 1.58; 1.73 × 10−4 | 1.58; 1.73 × 10−4 | 2.11; 4.54 × 10−8 | 1.58; 1.72 × 10−4 | 1.71; 2.20 × 10−5 | 1.71; 2.20 × 10−5 | |||
| Extracellular protein (EP) | 1.32; 0.0106 | 1.45; 1.36 × 10−3 | 1.58; 1.73 × 10−4 | 1.71; 2.20 × 10−5 | 1.85; 2.80 × 10−6 | 1.71; 2.20 × 10−5 | 1.85; 2.80 × 10−6 | 1.71; 2.20 × 10−5 | 1.58; 1.72 × 10−4 | ||
| Vacuolar protein (VP) | 1.19; 0.0835 | 1.19; 0.0835 | 1.06; 0.6560 | 1.32; 0.0106 | 1.45; 1.36 × 10−3 | 1.32; 0.0106 | 1.58; 1.72 × 10−4 | 1.32; 0.0106 | 1.32; 0.0106 | 1.45; 1.36 × 10−3 | |
| Lysosomal protein (LP) | 0.92; 5.15 | 1.32; 0.0106 | 1.32; 0.0106 | 1.32; 0.0106 | 1.19; 0.0835 | 1.32; 0.0106 | 1.32; 0.0106 | 1.45; 1.36 × 10−3 | 1.19; 0.0835 | 1.45; 1.36 × 10−3 | 1.19; 0.0835 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, G.; Tang, G.; Wang, C. Congruence Amidst Discordance between Sequence and Protein-Content Based Phylogenies of Fungi. J. Fungi 2020, 6, 134. https://doi.org/10.3390/jof6030134
Xiao G, Tang G, Wang C. Congruence Amidst Discordance between Sequence and Protein-Content Based Phylogenies of Fungi. Journal of Fungi. 2020; 6(3):134. https://doi.org/10.3390/jof6030134
Chicago/Turabian StyleXiao, Guohua, Guirong Tang, and Chengshu Wang. 2020. "Congruence Amidst Discordance between Sequence and Protein-Content Based Phylogenies of Fungi" Journal of Fungi 6, no. 3: 134. https://doi.org/10.3390/jof6030134