Genome Sequence Analysis of Auricularia heimuer Combined with Genetic Linkage Map

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Genomic DNA Extraction
2.3. DNA Library Construction and Illumina Deep Sequencing
2.4. Gene Assembly, Prediction, and Functional Annotation
2.5. Annotation of Genes Encoding Carbohydrate-Active Enzymes (CAZymes)
2.6. Phylogenomic Analysis of A. heimuer and Related Genomes
2.7. Analysis of Gene Families Expansion and Contraction
2.8. Preparation of Mating-type Mapping Population and Mapping
2.9. Analysis of Genomic Structure of the Mating-type Locus in A. heimuer
2.10. Gene Synteny Analysis of MAT-A of A. heimuer, A. subglabra, and E. glandulosa
3. Results
3.1. The Genome Features of A14-8
3.2. Lignocellulolysis
3.3. Gene Families Expansion and Contraction of CAZymes
3.4. Alcohol Dehydrogenases
3.5. Phylogenomic Analysis
3.6. Mating-Type Analysis of Mapping Population
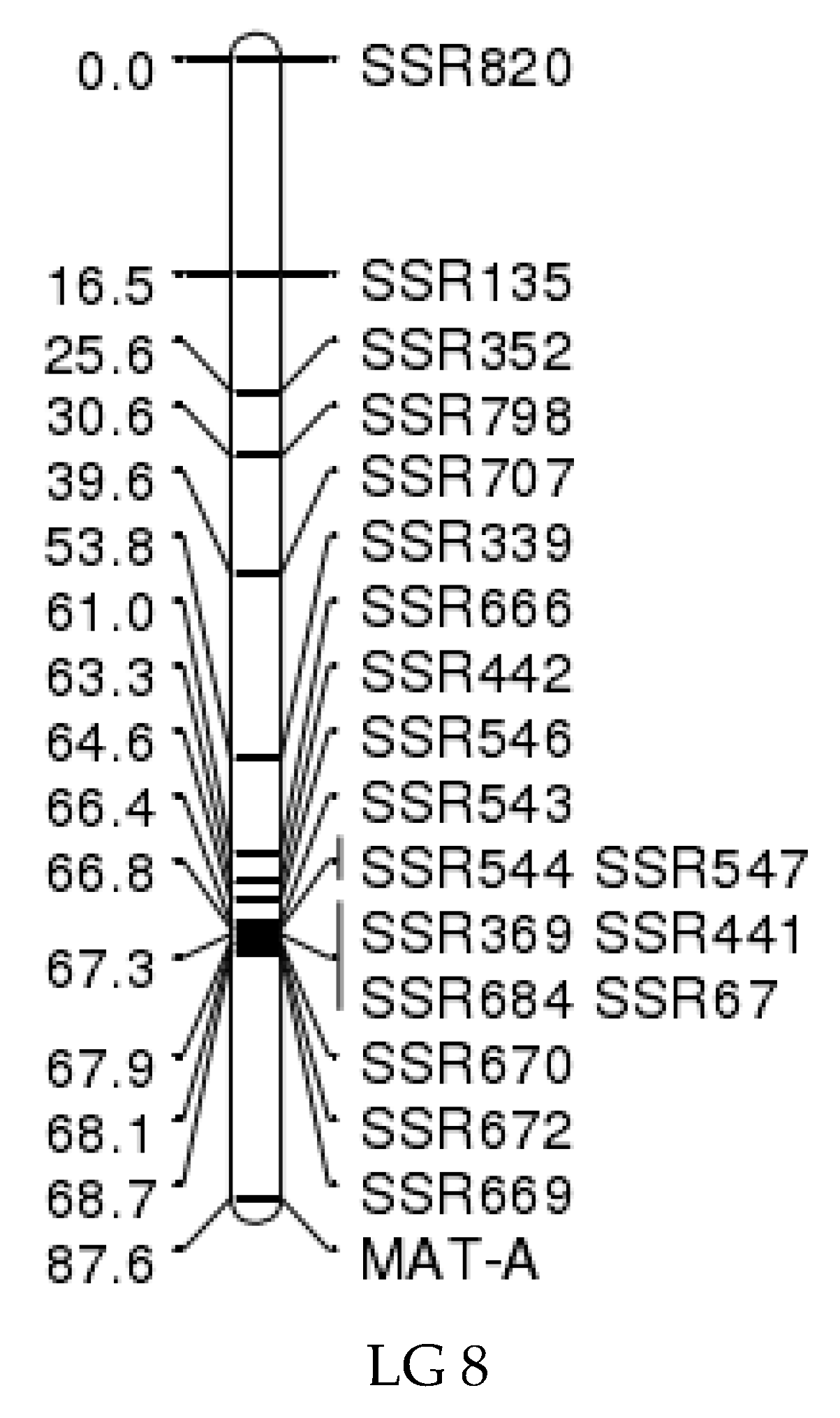
3.7. Mating-Type Location on the Genetic Linkage Map
3.8. Structure of the Mating-type Locus
3.9. Analysis of the Synteny of MAT-A in the A. heimuer, A. subglabra, and E. glandulosa Genomes
4. Discussion
4.1. Genome Features
4.2. CAZymes
4.3. Expansion and Contraction of CAZymes
4.4. Alcohol Dehydrogenases
4.5. Phylogenomic Analysis
4.6. Mating-type Locus of A. heimuer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yuan, Y.; Wu, F.; Si, J.; Zhao, Y.-F.; Dai, Y.-C. Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics 2018, 111, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Lou, L.H. Cultivation of Auricularia on Logs in China. In Tropical Mushrooms Biological Nature and Cultivation Methods; Chang, S.T., Quimio, T.H., Eds.; The Chinese University Press: Hong Kong, China, 1982; pp. 437–441. [Google Scholar]
- Ying, C.; Fang-jie, Y.; Gui-juan, L.; Hai-ying, W.; Yan, L. The Announcements and Suggestions of Auricularia auricula Cultivation Using Substitute Media. Edible Fungi China 2010, 29, 55–58. [Google Scholar]
- Yao, F.J.; Lu, L.X.; Wang, P.; Fang, M.; Zhang, Y.M.; Chen, Y.; Zhang, W.T.; Kong, X.H.; Lu, J.; Honda, Y. Development of a Molecular Marker for Fruiting Body Pattern in Auricularia auricula-judae. Mycobiology 2018, 46, 72–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang-jie, Y.; You-min, Z.; Ying, C. The Industrial Development Situation of Auricularia auricular of China. North. Hortic. 2010, 18, 209–211. [Google Scholar]
- Maehara, T.; Ichinose, H.; Furukawa, T.; Ogasawara, W.; Takabatake, K.; Kaneko, S. Ethanol production from high cellulose concentration by the basidiomycete fungus Flammulina velutipes. Fungal Biol-Uk 2013, 117, 220–226. [Google Scholar] [CrossRef]
- Okamura-Matsui, T.; Tomoda, T.; Fukuda, S.; Ohsugi, M. Discovery of alcohol dehydrogenase from mushrooms and application to alcoholic beverages. J. Mol. Catal. B Enzym. 2003, 23, 133–144. [Google Scholar] [CrossRef]
- Wu, F.; Yuan, Y.; Malysheva, V.F.; Du, P.; Dai, Y.-C. Species clarification of the most important and cultivated Auricularia mushroom “Heimuer”: Evidence from morphological and molecular data. Phytotaxa 2014, 186, 241–253. [Google Scholar] [CrossRef]
- Morin, E.; Kohler, A.; Baker, A.R.; Foulongne-Oriol, M.; Lombard, V.; Nagy, L.G.; Ohm, R.A.; Patyshakuliyeva, A.; Brun, A.; Aerts, A.L.; et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA 2012, 109, 17501–17506. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.; Salamov, A.A.; Brown, D.W.; Nagy, L.G.; Floudas, D.; Held, B.W.; Levasseur, A.; Lombard, V.; Morin, E.; Otillar, R.; et al. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc. Natl. Acad. Sci. USA 2014, 111, 9923–9928. [Google Scholar] [CrossRef] [Green Version]
- Ohm, R.A.; de Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; de Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 28, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Stajich, J.E.; Wilke, S.K.; Ahrén, D.; Au, C.H.; Birren, B.W.; Borodovsky, M.; Burns, C.; Canbäck, B.; Casselton, L.A.; Cheng, C.K.; et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. USA 2010, 107, 11889–11894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehouse, H. Multiple-allelomorph heterothallism in the fungi. New Phytol. 1949, 48, 212–244. [Google Scholar] [CrossRef]
- Lu, L.-X.; Yao, F.-J.; Wang, P.; Fang, M.; Zhang, Y.-M.; Zhang, W.-T.; Kong, X.-H.; Lu, J. Construction of a genetic linkage map and QTL mapping of agronomic traits in Auricularia auricula-judae. J. Microbiol. 2017, 55, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome. Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome. Biol. 2006, 7, S10. [Google Scholar] [CrossRef] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Cao, J.; Junkai, H.; Wang, Z.; Guo, Z.; Chen, Y.; Ma, S.; Liu, J. Genome sequencing and comparative genomics reveal the potential pathogenic mechanism of Cercospora sojina Hara on soybean. DNA Res. 2017, 25, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, R.M.; Tegenfeldt, F.; Li, J.; Zdobnov, E.M.; Kriventseva, E.V. OrthoDB: A hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013, 41, D358–D365. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Han, M.; Thomas, G.; Lugo-Martinez, J.; Hahn, M. Estimating Gene Gain and Loss Rates in the Presence of Error in Genome Assembly and Annotation Using CAFE 3. Mol. Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 2011, 93, 343–349. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salamov, A.A.; Solovyev, V.V. Ab initio Gene Finding in Drosophila Genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radakovits, R.; Jinkerson, R.E.; Fuerstenberg, S.I.; Tae, H.; Settlage, R.E.; Boore, J.L.; Posewitz, M.C. Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropsis gaditana. Nat Commun. 2012, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic Origin of Enzymatic Lignin Decomposition Reconstructed from 31 Fungal Genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, H.; Umezawa, K.; Takeda, K.; Sugimoto, N.; Ishida, T.; Samejima, M.; Ohno, H.; Yoshida, M.; Igarashi, K.; Nakamura, N. Discovery of a Eukaryotic Pyrroloquinoline Quinone-Dependent Oxidoreductase Belonging to a New Auxiliary Activity Family in the Database of Carbohydrate-Active Enzymes. PLoS ONE 2014, 9, e104851. [Google Scholar] [CrossRef] [Green Version]
- De Smidt, O.; Preez, J.C.D.; Albertyn, J. The alcohol dehydrogenases of Saccharomyces cerevisiae: A comprehensive review. Fems. Yeast Res. 2010, 8, 967–978. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.S.; Park, H.R.; Yoon, D.E.; Nam, J.Y.; et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef]
- Hatakka, A.; Hammel, K.E. Fungal biodegradation of lignocelluloses. In Industrial Applications; Hofrichter, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 319–340. [Google Scholar]
- Van Peer, A.F.; Park, S.Y.; Shin, P.G.; Jang, K.Y.; Yoo, Y.B.; Park, Y.J.; Lee, B.M.; Sung, G.H.; James, T.Y.; Kong, W.S. Comparative genomics of the mating-type loci of the mushroom Flammulina velutipes reveals widespread synteny and recent inversions. PLoS ONE 2011, 6, e22249. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G.; et al. Genome Sequence of the Edible Cultivated Mushroom Lentinula edodes (Shiitake) Reveals Insights into Lignocellulose Degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.; Gong, M.; Zheng, H.; Chen, M.; Zhang, L.; Wang, H.; Jiang, J.; Wu, L.; Zhu, Y.; Zhu, G.; et al. Sequencing and Comparative Analysis of the Straw Mushroom (Volvariella volvacea) Genome. PLoS ONE 2013, 8, e58294. [Google Scholar] [CrossRef]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiezynski, K.M.; Day, P.R. Heterokaryon formation in Coprinus lagopus. Genet. Res. Camb. 1960, 1, 114–128. [Google Scholar] [CrossRef]
- Fraser, J.A.; Diezmann, S.; Subaran, R.L.; Allen, A.; Lengeler, K.B.; Dietrich, F.S.; Heitman, J. Convergent Evolution of Chromosomal Sex-Determining Regions in the Animal and Fungal Kingdoms. PLoS Biol. 2004, 2, 2243–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkeren, G.; Kronstad, J.W. Linkage of mating-type loci distinguishes bipolar from tetrapolar mating in basidiomycetous smut fungi. Proc. Natl. Acad. Sci. USA 1994, 91, 7085–7089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.; Bakkeren, G.; Wong, K.; Sherwood, J.E.; Kronstad, J.W. The mating-type and pathogenicity locus of the fungus Ustilago hordei spans a 500-kb region. Proc. Natl. Acad. Sci. USA 1999, 96, 15026–15031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, T.Y.; Srivilai, P.; Kües, U.; Vilgalys, R. Evolution of the bipolar mating system of the mushroom Coprinellus disseminatus from its tetrapolar ancestors involves loss of mating-type-specific pheromone receptor function. Genetics 2006, 172, 1877–1891. [Google Scholar] [CrossRef] [Green Version]
- Yi, R.; Tachikawa, T.; Mukaiyama, H.; Mochida, Y.; Ishikawa, M.; Aimi, T. DNA-mediated transformation system in a bipolar basidiomycete, Pholiota microspora (P. nameko). Mycoscience 2009, 50, 123–129. [Google Scholar] [CrossRef]
- James, T.Y.; Lee, M.; van Diepen, L.T.A. A Single Mating-Type Locus Composed of Homeodomain Genes Promotes Nuclear Migration and Heterokaryosis in the White-Rot Fungus Phanerochaete chrysosporium. Eukaryot Cell 2011, 10, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Au, C.H.; Wong, M.C.; Bao, D.; Zhang, M.; Song, C.; Song, W.; Law, P.T.W.; Kües, U.; Kwan, H.S. The genetic structure of the A mating-type locus of Lentinula edodes. Gene 2014, 535, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Casselton, L.A.; Asanteowusu, R.N.; Banham, A.H.; Kingsnorth, C.S.; Kües, U.; O’Shea, S.F.; Pardo, E.H. Mating type control of sexual development in Coprinus cinereus. Can. J. Bot. 1995, 73, 266–272. [Google Scholar] [CrossRef]
- Specht, C.A.; Stankis, M.M.; Giasson, L.; Novotny, C.P.; Ullrich, R.C. Functional analysis of the homeodomain-related proteins of the A alpha locus of Schizophyllum commune. Proc. Natl. Acad. Sci. USA 1992, 89, 7174–7178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhilin, L.; Ruili, Z. Advances of Genomics-assisted Cultivation and Breedingof Edible and Medicinal Mushrooms. Acta Edulis Fungi 2018, 25, 93–106. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Features. | Properties of Predicted Gene Models | ||
|---|---|---|---|
| Genome size (Mb) | 43.6 | KEGG alignment | 2257 |
| GC content (%) | 57.09 | Interpro signature | 9254 |
| Scaffolds N50 (bp) | 276,651 | Pfam alignment | 12,279 |
| Number of scaffolds | 535 | CAZyme alignment | 509 |
| Number of predict gene models | 14,094 | Signal peptide | 1694 |
| Average of gene length (bp) | 1737.54 | Transmembrane domain | 2441 |
| Average of cds length (bp) | 1390 | ||
| Average of exon number | 5.71 | ||
| Average of intron number | 4.71 | ||
| Average of exon length (bp) | 244 | ||
| Average of intron length (bp) | 74 | ||
| Repetitive sequences (%) | 3.73 | ||
| CAZy Family | A.hei | T.fuc | A.sub | L.edo | F.vel | P.ost | S.com | G.luc | S.lac | C.cin | A.bis | T.mes | V.vol |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CBM | 60 | 11 | 56 | 30 | 22 | 54 | 20 | 28 | 12 | 71 | 29 | 10 | 66 |
| CE | 18 | 14 | 18 | 11 | 19 | 14 | 12 | 8 | 7 | 23 | 18 | 8 | 11 |
| GH | 265 | 107 | 292 | 209 | 190 | 212 | 206 | 205 | 125 | 178 | 152 | 56 | 195 |
| GT | 51 | 53 | 51 | 62 | 57 | 48 | 54 | 52 | 37 | 51 | 41 | 50 | 51 |
| PL | 11 | 2 | 11 | 3 | 20 | 18 | 11 | 6 | 2 | 6 | 4 | 2 | 19 |
| AA | 104 | 14 | 124 | 85 | 91 | 133 | 78 | 99 | 41 | 124 | 90 | 8 | 118 |
| sum | 509 | 201 | 552 | 400 | 399 | 479 | 381 | 398 | 224 | 453 | 334 | 134 | 460 |
| Substate | CAZy Family | Species | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A. hei. | A.sub | L. edo | F. vel | P. ost | S. com. | S. lac. | G. luc | A. bis | V. vol | L. bic | C. cin. | ||
| Crystalline cellulose | CBM1 | 43 | 43 | 23 | 15 | 31 | 4 | 8 | 20 | 16 | 54 | 1 | 50 |
| GH6 | 2 | 2 | 1 | 2 | 3 | 1 | 1 | 1 | 1 | 4 | 0 | 5 | |
| GH7 | 5 | 6 | 3 | 3 | 16 | 2 | 0 | 3 | 1 | 12 | 0 | 6 | |
| AA9 | 19 | 20 | 13 | 19 | 28 | 22 | 5 | 15 | 11 | 31 | 3 | 34 | |
| sum | 69 | 71 | 40 | 39 | 78 | 29 | 14 | 39 | 29 | 101 | 4 | 95 | |
| Lignin | AA1_1 | 0 | 0 | 14 | 8 | 11 | 2 | 4 | 13 | 11 | 11 | 10 | 17 |
| AA1_2 | 1 | 1 | 1 | 2 | 1 | 0 | 2 | 1 | 1 | 0 | 4 | 0 | |
| AA2 | 21 | 21 | 10 | 3 | 9 | 2 | 1 | 10 | 5 | 9 | 3 | 4 | |
| AA3_1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | |
| AA3_2 | 22 | 34 | 17 | 20 | 36 | 15 | 7 | 26 | 28 | 26 | 8 | 27 | |
| AA3_3 | 3 | 6 | 4 | 4 | 4 | 4 | 5 | 4 | 4 | 4 | 2 | 2 | |
| AA3_4 | 1 | 3 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| AA4 | 1 | 1 | 1 | 3 | 1 | 2 | 2 | 1 | 1 | 1 | 0 | 0 | |
| AA5_1 | 5 | 7 | 5 | 7 | 15 | 2 | 3 | 10 | 8 | 4 | 10 | 6 | |
| AA6 | 4 | 4 | 2 | 3 | 2 | 4 | 2 | 2 | 4 | 2 | 2 | 3 | |
| AA7 | 15 | 14 | 11 | 19 | 23 | 12 | 8 | 12 | 13 | 26 | 7 | 18 | |
| AA8 | 9 | 11 | 2 | 3 | 1 | 1 | 2 | 1 | 5 | 1 | 0 | 3 | |
| sum | 83 | 102 | 68 | 73 | 103 | 45 | 36 | 80 | 81 | 84 | 46 | 81 | |
| Strain | Mating-Type Locus | Gene | Length of Amino Acid (aa) | Subunit | The Gap in the Same Subunit (bp) | The Gap Between α and β Subunit (kb) |
| A14-8 | scaffold45 | HD1-1(MN267022) | 683 | α subunit | 153 | 36.725 |
| HD1-2(MN267023) | 626 | |||||
| A14-5 | contig976 | HD1-1(MN267026) | 593 | α subunit | 294 | 36.716 |
| HD1-2(MN267027) | 683 | |||||
| 18-119 | contig313 | HD1-1(MN267028) | 684 | α subunit | 1143 | 37.563 |
| HD1-2(MN267029) | 633 | |||||
| Dai 13782 | NEKD01000007.1 | HD1-1(MN442077) | 615 | α subunit | 171 | 41.16 |
| HD1-2(MN442078) | 613 | |||||
| Strain | Mating-type Locus | Gene | Length of Amino Acid (aa) | Subunit | The Gap in the Same Subunit (bp) | |
| A14-8 | scaffold45 | HD2-1(MN267021) | 693 | β subunit | 2198 | |
| HD2-2(MN267024) | 628 | |||||
| A14-5 | contig976 | HD2-1(MN267030) | 628 | β subunit | 153 | |
| HD2-2(MN267031) | 626 | |||||
| 18-119 | contig313 | HD2-1(MN267032) | 622 | β subunit | 411 | |
| HD2-2(MN267033) | 650 | |||||
| Dai 13782 | NEKD01000007.1 | HD2-1(MN442079) | 715 | β subunit | 145 | |
| HD2-2(MN442080) | 599 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, M.; Wang, X.; Chen, Y.; Wang, P.; Lu, L.; Lu, J.; Yao, F.; Zhang, Y. Genome Sequence Analysis of Auricularia heimuer Combined with Genetic Linkage Map. J. Fungi 2020, 6, 37. https://doi.org/10.3390/jof6010037
Fang M, Wang X, Chen Y, Wang P, Lu L, Lu J, Yao F, Zhang Y. Genome Sequence Analysis of Auricularia heimuer Combined with Genetic Linkage Map. Journal of Fungi. 2020; 6(1):37. https://doi.org/10.3390/jof6010037
Chicago/Turabian StyleFang, Ming, Xiaoe Wang, Ying Chen, Peng Wang, Lixin Lu, Jia Lu, Fangjie Yao, and Youmin Zhang. 2020. "Genome Sequence Analysis of Auricularia heimuer Combined with Genetic Linkage Map" Journal of Fungi 6, no. 1: 37. https://doi.org/10.3390/jof6010037