
What Aspects of Phenotype Determine Risk for Sudden Cardiac Death in Pediatric Hypertrophic Cardiomyopathy?

Abstract
:1. Introduction
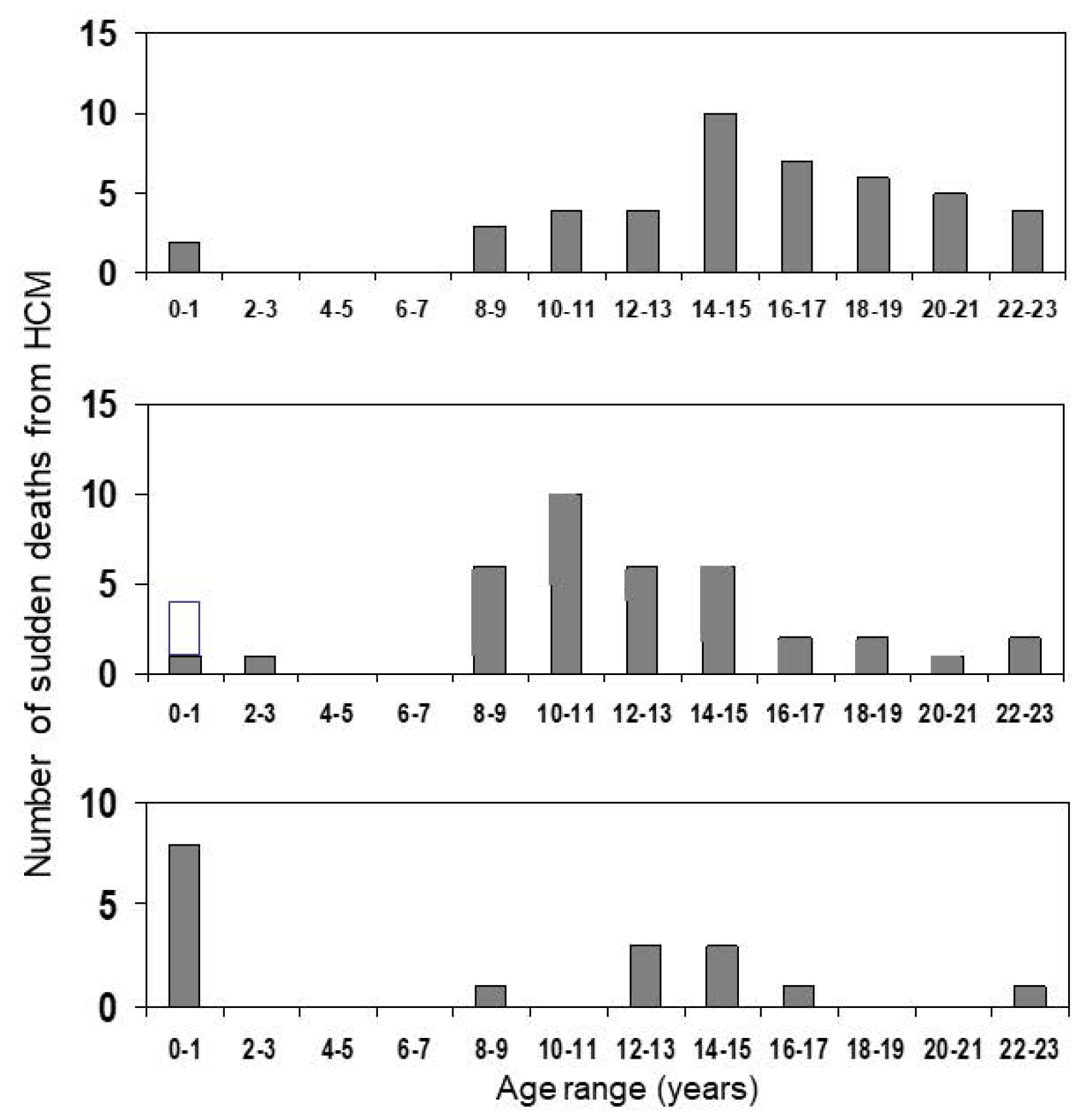
2. At What Age Is the Risk Highest?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Era | Age D (y) | Setting (n) | FU (y) | Patient yrs | Events (Type) | “Annual” Event rate (%) | Incl. Comm. SCD? | Exclusions | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1962–1980 | 9 | SC, TC (37) | 9 | 333 | 18 (SCD) | 4.8 | No | FH of SCD | McKenna et al., 1984 [21] |
| 1958–1997 | 5.0 | SC, TC (99) | 4.8 | 475 | 12 (SCD) | 2.5 | No | None | Yetman et al., 1998 [22] |
| 1968–1998 | 6.3 | MC, RC (66) | 12.0 | 789 | 10 (SCD) | Total:1.3 HDBB = 0 NST: ns-HCM = 2.3 RAS = 2.3 | No | None | Östman-Smith et al., 1999 [23] |
| 1970–2003 | 5.7 | MC, RC (128) | ns:10.9 RAS:12.0 | Ns = 948 RAS = 492 | Ns = 12 RAS = 4 (SCD) | ns-HCM = 1.3 RAS = 0.8 | No | None | Östman-Smith et al., 2005 [9] |
| 1972–2004 | 4.6 | MC, Nat Cohort (150) | 7.0 | 1050 | 39 (under FU = 27) (LAE) | Total incl cSCD: 3.7 FU-grp = 2.0 | Yes | None | Östman-Smith et al., 2008 [12] |
| 1985–2006 | 10.6 | SC, TC (96) | 6.4 | 614 | 3 (SCD) | 0.5 | No | None | Decker et al., 2009 [24] |
| 1980–2001 | ?(14.4 at EP-study) | SC, TC (131) | 6.4 | 838 | 22 (MACE) | 2.6 | No | Prev MACE | Moak et al., 2011 [25] |
| 1993–2014 | 14.1 | SC, TC (112) | 6.5 | 728 | 13 (LAE) | 1.8 | No | None, phenocopies included | Ziolkowska et al., 2015 [26] |
| 1987–1996 | 0.45 | MC, nat Cohort (80) | 14.0 | 1120 | 4 | 0.4 | No | Patients > 10 yr at diagnosis | Bharucha et al., 2015 [27] |
| 1972–2014 | 8.4 | MC, nat Cohort (155) | 10.9 | 1766 | 39 (Under FU = 27) | Total incl cSCD: 2.4 FU-grp: <1999:1.8 ≥1999-:1.1 | Yes | None | Östman-Smith et al., 2017 [19] |
| 1974–2016 | 12.2 | SC, TC (100) | 9.2 | 920 | 19 (LAE) | 2.1 | No | RAS-HCM | Maurizi et al., 2018 [20] |
| 1970–2017 | 11 | MC, TC (1024) | 5.3 | 5984 | 89 (MACE) | 1.5 | No | RAS-HCM | Norrish et al., 2019 [28] |
| ?–2017 | 9.8 | MC, TC (572) | 5.0 | 2855 | 53 (LAE) | 1.9 | No | RAS-HCM (PRIMaCY) | Miron et al., 2020 [18] |
| ?–2017 | 13.8 | MC, TC (285) | 4.9 | 1400 | 22 (LAE) | 1.6 | No | RAS-HCM (ShaRe) | Miron et al., 2020 [18] |
| 1972–2016 | 10.9 | MC, Nat Cohort (151) | 11.6 | 2008 | ns-HCM = 27 RAS-HCM = 6(LAE) | ns-HCM = 1.8 RAS = 1.7 | No | Patients presenting with arrest | Östman-Smith et al., 2021 [29] |
3. Sex
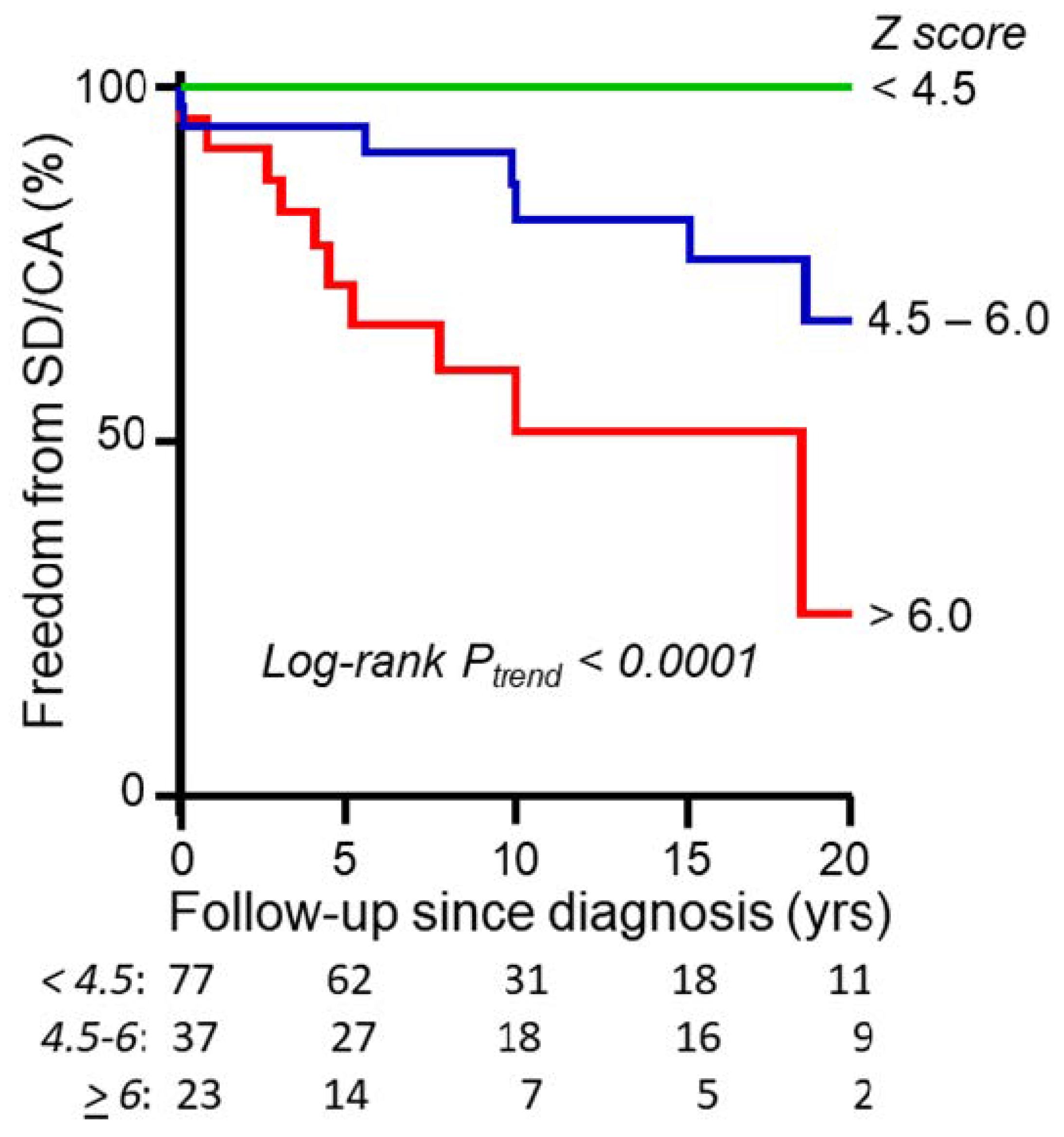
4. Left Ventricular Hypertrophy
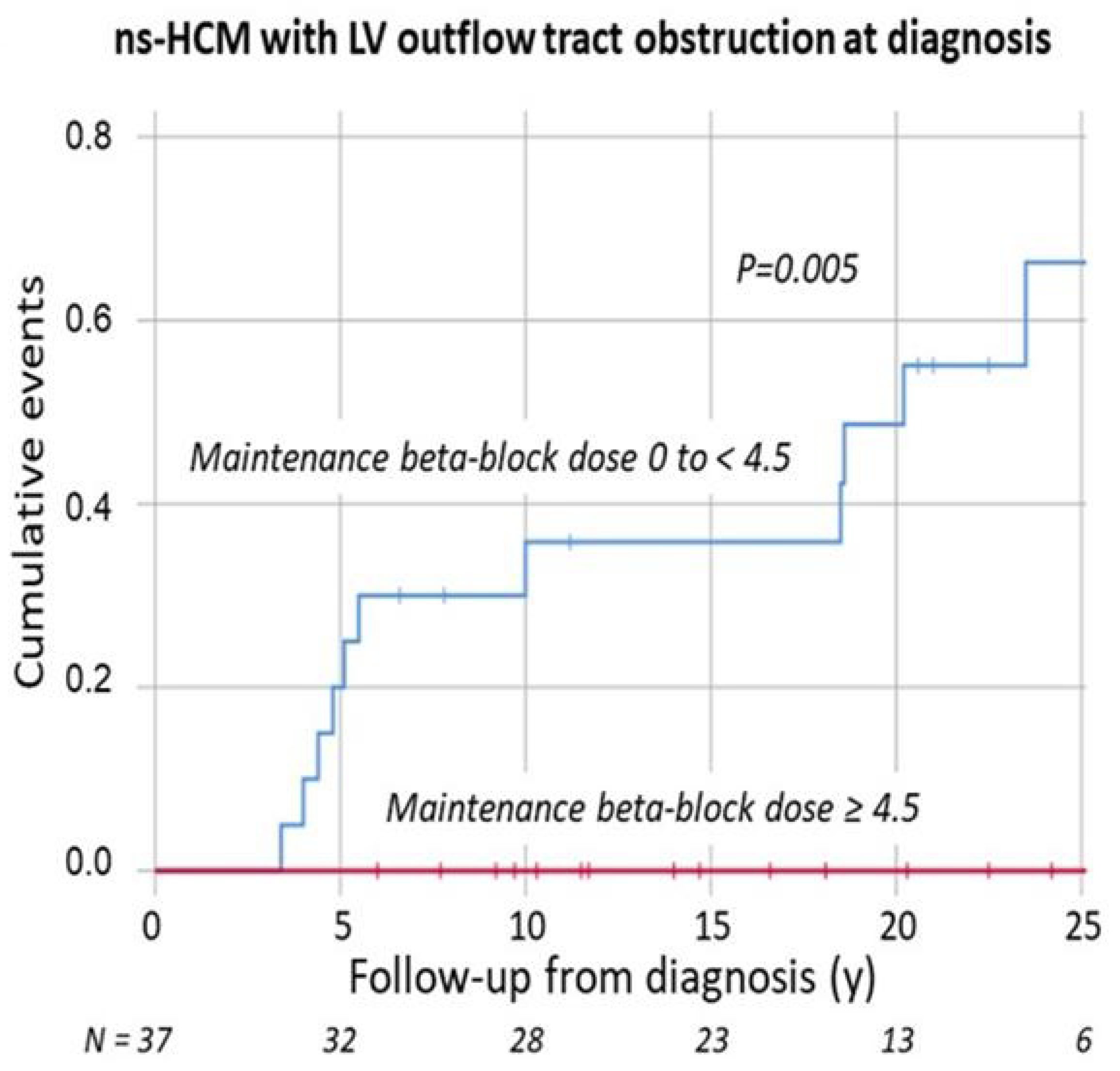
5. Left Ventricular Outflow Tract Obstruction
6. Diastolic Myocardial Function—Restrictive Physiology
7. Left Atrial Enlargement
8. Electrocardiographic Phenotype
9. Myocyte Energy Deficit, and Its Importance for Ischaemia on Exercise
10. Myocardial Fibrosis and Scarring
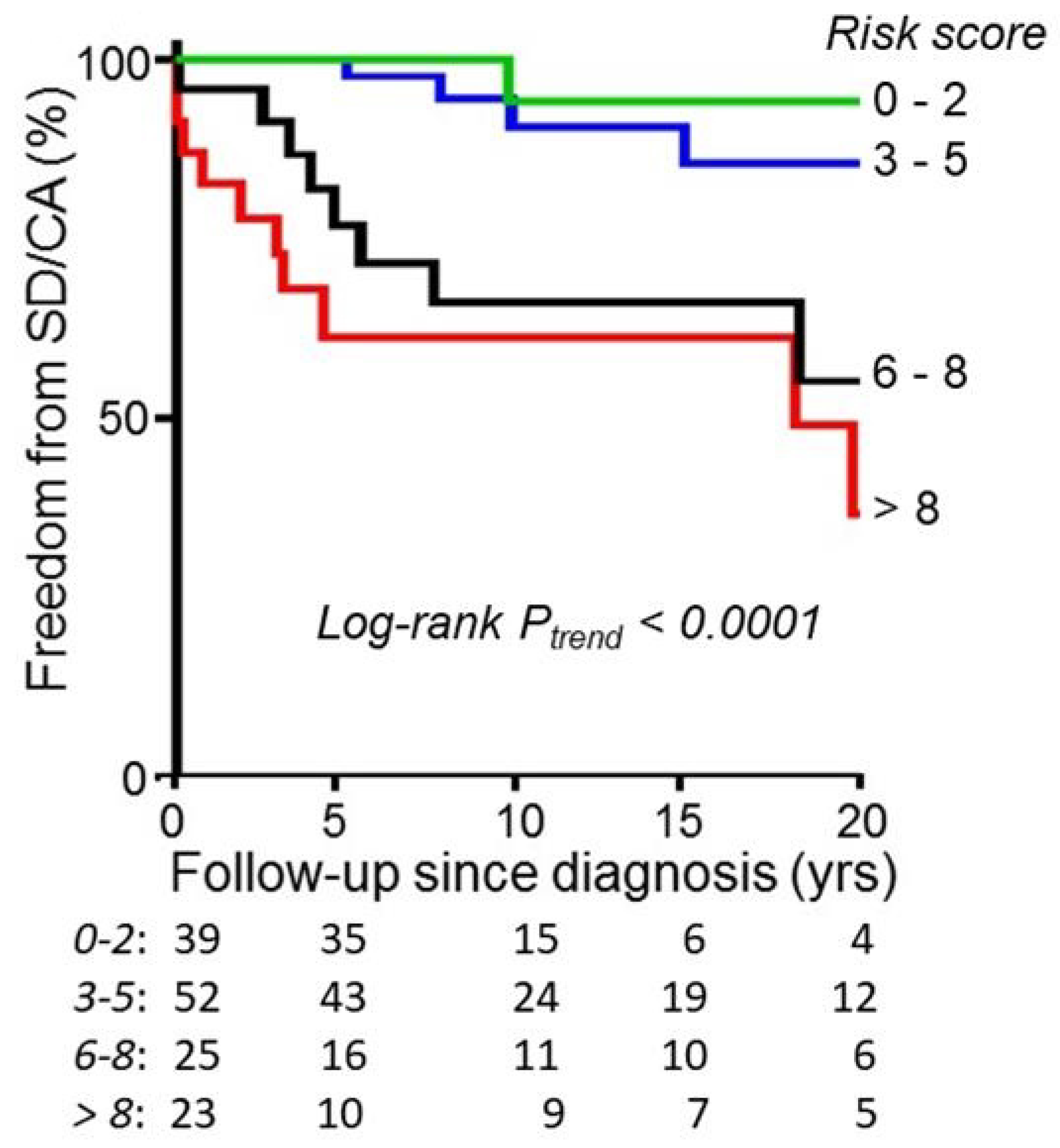
11. Risk-Assessment Algorithms
12. Is Phenotype or Genotype More Important in Determining Risk?
13. Discussion
13.1. Current Knowledge Gaps
13.2. Future Directions
14. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [PubMed]
- Reneman, R.; Schmitz, J.; Snoeckx, L.; Lambregts, J. Differences in the echocardiographic dimensions of the heart between females and males. In Echocardiology; Lancée, C., Ed.; Martinus Nijhoff: The Hague, The Netherlands, 1979; pp. 53–60. [Google Scholar]
- Grandi, A.M.; Venco, A.; Barzizza, F.; Scalise, F.; Pantaleo, P.; Finardi, G. Influence of age and sex on left ventricular anatomy and function in normals. Cardiology 1992, 81, 8–13. [Google Scholar] [CrossRef]
- Hada, Y.; Sakamoto, T.; Amano, K.; Yamaguchi, T.; Takenaka, K.; Takahashi, H.; Takikawa, R.; Hasegawa, I.; Takahashi, T.; Suzuki, J.; et al. Prevalence of hypertrophic cardiomyopathy in a population of adult Japanese workers as detected by echocardiographic screening. Am. J. Cardiol. 1987, 59, 183–184. [Google Scholar] [CrossRef]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: Echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Arola, A.; Jokinen, E.; Ruuskanen, O.; Saraste, M.; Pesonen, E.; Kuusela, A.L.; Tikanoja, T.; Paavilainen, T.; Simell, O. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am. J. Epidemiol. 1997, 146, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Nugent, A.; Daubeney, P.; Chondros, P.; Carlin, J.; Cheung, M.; Wilkinson, L.; Davis, A.; Kahler, S.; Chow, C.; Wilkinson, J.; et al. The epidemiology of childhood cardiomyopathy in Australia. N. Engl. J. Med. 2003, 348, 1639–1646. [Google Scholar] [CrossRef]
- McKenna, W.; Deanfield, J.; Faruqui, A.; England, D.; Oakley, C.; Goodwin, J. Prognosis in hypertrophic cardiomyopathy: Role of age and clinical, electrocardiographic and hemodynamic features. Am. J. Cardiol. 1981, 47, 532–538. [Google Scholar] [CrossRef]
- Östman-Smith, I.; Wettrell, G.; Keeton, B.; Riesenfeld, T.; Holmgren, D.; Ergander, U. Echocardiographic and electrocardiographic identification of those children with hypertrophic cardiomyopathy who should be considered at high-risk of dying suddenly. Cardiol. Young. 2005, 15, 632–642. [Google Scholar] [CrossRef]
- Norrish, G.; Field, E.; McLeod, K.; Ilina, M.; Stuart, G.; Bhole, V.; Uzun, O.; Brown, E.; Daubeney, P.E.F.; Lota, A.; et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: A retrospective study in United Kingdom. Eur. Heart J. 2019, 40, 986–993. [Google Scholar] [CrossRef]
- Marston, N.A.; Han, L.; Olivotto, I.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Ingles, J.; Semsarian, C.; Jacoby, D.; et al. Clinical characteristics and outcomes in childhood-onset hypertrophic cardiomyopathy. Eur. Heart J. 2021, 42, 1988–1996. [Google Scholar] [CrossRef]
- Östman-Smith, I.; Wettrell, G.; Keeton, B.; Holmgren, D.; Ergander, U.; Gould, S.; Bowker, C.; Verdicchio, M. Age- and gender-specific mortality rates in childhood hypertrophic cardiomyopathy. Eur. Heart J. 2008, 29, 1160–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugishita, Y.; Matsuda, M.; Iida, K.; Koshinaga, J.; Ueno, M. Sudden cardiac death at exertion. Jpn. Circ. J. 1983, 47, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Carney, K.P.; Lever, H.M.; Lewis, J.F.; Barac, I.; Casey, S.A.; Sherrid, M.V. Relationship of race to sudden cardiac death in competitive athletes with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2003, 41, 974–980. [Google Scholar] [CrossRef] [Green Version]
- Östman-Smith, I.; Rossano, J.W.; Shaddy, R.E. Hypertrophic cardiomyopathy: Do sudden death prevention strategies in children differ between Europe and North America? Curr. Opin. Cardiol. 2013, 28, 130–138. [Google Scholar] [CrossRef]
- Maron, B.J.; Roberts, W.C.; Epstein, S. Sudden death in hypertrophic cardiomyopathy: A profile of 78 patients. Circulation 1982, 65, 1388–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colan, S.D.; Lipshultz, S.E.; Lowe, A.M.; Sleeper, L.A.; Messere, J.; Cox, G.F.; Lurie, P.R.; Orav, E.J.; Towbin, J.A. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Circulation 2007, 115, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Miron, A.; Lafreniere-Roula, M.; Steve Fan, C.P.; Armstrong, K.R.; Dragulescu, A.; Papaz, T.; Manlhiot, C.; Kaufman, B.; Butts, R.J.; Gardin, L.; et al. A Validated Model for Sudden Cardiac Death Risk Prediction in Pediatric Hypertrophic Cardiomyopathy. Circulation 2020, 142, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Östman-Smith, I.; Sjöberg, G.; Rydberg, A.; Larsson, P.; Fernlund, E. Predictors of risk for sudden death in childhood hypertrophic cardiomyopathy: The importance of the ECG risk score. Open Heart 2017, 4, e000658. [Google Scholar] [CrossRef]
- Maurizi, N.; Passantino, S.; Spaziani, G.; Girolami, F.; Arretini, A.; Targetti, M.; Pollini, I.; Tomberli, A.; Pradella, S.; Calabri, G.B.; et al. Long-term Outcomes of Pediatric-Onset Hypertrophic Cardiomyopathy and Age-Specific Risk Factors for Lethal Arrhythmic Events. JAMA Cardiol. 2018, 3, 520–525. [Google Scholar] [CrossRef] [Green Version]
- McKenna, W.J.; Deanfield, J.E. Hypertrophic cardiomyopathy: An important cause of sudden death. Arch. Dis. Child. 1984, 59, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Yetman, A.T.; Hamilton, R.M.; Benson, L.N.; McCrindle, B.W. Long-term outcome and prognostic determinants in children with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1998, 32, 1943–1950. [Google Scholar] [CrossRef] [Green Version]
- Östman-Smith, I.; Wettrell, G.; Riesenfeld, T. A cohort study of childhood hypertrophic cardiomyopathy: Improved survival following high-dose beta-adrenoceptor antagonist treatment. J. Am. Coll. Cardiol. 1999, 34, 1813–1822. [Google Scholar] [CrossRef] [Green Version]
- Decker, J.A.; Rossano, J.W.; Smith, E.O.; Cannon, B.; Clunie, S.K.; Gates, C.; Jefferies, J.L.; Kim, J.J.; Price, J.F.; Dreyer, W.J.; et al. Risk factors and mode of death in isolated hypertrophic cardiomyopathy in children. J. Am. Coll. Cardiol. 2009, 54, 250–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moak, J.P.; Leifer, E.S.; Tripodi, D.; Mohiddin, S.A.; Fananapazir, L. Long-term follow-up of children and adolescents diagnosed with hypertrophic cardiomyopathy: Risk factors for adverse arrhythmic events. Pediatr. Cardiol. 2011, 32, 1096–1105. [Google Scholar] [CrossRef]
- Ziolkowska, L.; Turska-Kmiec, A.; Petryka, J.; Kawalec, W. Predictors of Long-Term Outcome in Children with Hypertrophic Cardiomyopathy. Pediatr. Cardiol. 2016, 37, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Bharucha, T.; Lee, K.J.; Daubeney, P.E.; Nugent, A.W.; Turner, C.; Sholler, G.F.; Robertson, T.; Justo, R.; Ramsay, J.; Carlin, J.B.; et al. Sudden death in childhood cardiomyopathy: Results from a long-term national population-based study. J. Am. Coll. Cardiol. 2015, 65, 2302–2310. [Google Scholar] [CrossRef] [Green Version]
- Norrish, G.; Ding, T.; Field, E.; Ziolkowska, L.; Olivotto, I.; Limongelli, G.; Anastasakis, A.; Weintraub, R.; Biagini, E.; Ragni, L.; et al. Development of a Novel Risk Prediction Model for Sudden Cardiac Death in Childhood Hypertrophic Cardiomyopathy (HCM Risk-Kids). JAMA Cardiol. 2019, 4, 918–927. [Google Scholar] [CrossRef]
- Östman-Smith, I.; Sjöberg, G.; Alenius Dahlqvist, J.; Larsson, P.; Fernlund, E. Sudden cardiac death in childhood hypertrophic cardiomyopathy is best predicted by a combination of electrocardiogram risk-score and HCMRisk-Kids score. Acta Paediatr. 2021, 110, 3105–3115. [Google Scholar] [CrossRef]
- Shaw, A.C.; Kalidas, K.; Crosby, A.H.; Jeffery, S.; Patton, M.A. The natural history of Noonan syndrome: A long-term follow-up study. Arch. Dis. Child. 2007, 92, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, G.; Pacileo, G.; Marino, B.; Digilio, M.C.; Sarkozy, A.; Elliott, P.; Versacci, P.; Calabro, P.; De Zorzi, A.; Di Salvo, G.; et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am. J. Cardiol. 2007, 100, 736–741. [Google Scholar] [CrossRef]
- Charron, P.; Carrier, L.; Dubourg, O.; Tesson, F.; Desnos, M.; Richard, P.; Bonne, G.; Guicheney, P.; Hainque, B.; Bouhour, J.B.; et al. Penetrance of familial hypertrophic cardiomyopathy. Genet. Couns. 1997, 8, 107–114. [Google Scholar] [PubMed]
- Terauchi, Y.; Kubo, T.; Baba, Y.; Hirota, T.; Tanioka, K.; Yamasaki, N.; Furuno, T.; Kitaoka, H. Gender differences in the clinical features of hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J. Cardiol. 2015, 65, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, Y.; Miyazaki, A.; Tsuda, E.; Hayama, Y.; Negishi, J.; Ohuchi, H. Electrocardiographic changes and long-term prognosis of children diagnosed with hypertrophic cardiomyopathy by the school screening program for heart disease in Japan. J. Cardiol. 2020, 75, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Spirito, P.; Bellone, P.; Harris, K.M.; Bernabo, P.; Bruzzi, P.; Maron, B.J. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N. Engl. J. Med. 2000, 342, 1778–1785. [Google Scholar] [CrossRef]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 2011, 58, e212–e260. [Google Scholar]
- Balaji, S.; DiLorenzo, M.P.; Fish, F.A.; Etheridge, S.P.; Aziz, P.F.; Russell, M.W.; Tisma, S.; Pflaumer, A.; Sreeram, N.; Kubus, P.; et al. Risk factors for lethal arrhythmic events in children and adolescents with hypertrophic cardiomyopathy and an implantable defibrillator: An international multicenter study. Heart Rhythm. 2019, 16, 1462–1467. [Google Scholar] [CrossRef]
- Pettersen, M.D.; Du, W.; Skeens, M.E.; Humes, R.A. Regression equations for calculation of z scores of cardiac structures in a large cohort of healthy infants, children, and adolescents: An echocardiographic study. J. Am. Soc. Echocardiogr. 2008, 21, 922–934. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar]
- Semsarian, C.; French, J.; Trent, R.J.; Richmond, D.R.; Jeremy, R.W. The natural history of left ventricular wall thickening in hypertrophic cardiomyopathy. Aust. N. Z. J. Med. 1997, 27, 51–58. [Google Scholar] [CrossRef]
- Maron, M.S.; Olivotto, I.; Betocchi, S.; Casey, S.A.; Lesser, J.R.; Losi, M.A.; Cecchi, F.; Maron, B.J. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N. Engl. J. Med. 2003, 348, 295–303. [Google Scholar] [CrossRef]
- Geske, J.B.; Ong, K.C.; Siontis, K.C.; Hebl, V.B.; Ackerman, M.J.; Hodge, D.O.; Miller, V.M.; Nishimura, R.A.; Oh, J.K.; Schaff, H.V.; et al. Women with hypertrophic cardiomyopathy have worse survival. Eur. Heart J. 2017, 38, 3434–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javidgonbadi, D.; Andersson, B.; Abdon, N.J.; Schaufelberger, M.; Östman-Smith, I. Factors influencing long-term heart failure mortality in patients with obstructive hypertrophic cardiomyopathy in Western Sweden: Probable dose-related protection from beta-blocker therapy. Open Heart 2019, 6, e000963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Liu, P.Y.; Lin, L.J.; Chen, J.H.; Tsai, L.M. Clinical characteristics and outcomes of hypertrophic cardiomyopathy in Taiwan--a tertiary center experience. Clin. Cardiol. 2007, 30, 177–182. [Google Scholar] [CrossRef]
- El Saiedi, S.; Behairy, N.H.; Kharabish, A.; Esmail, R.; Seliem, Z.S.; Shafik, M.; El Mozy, W. Delayed Myocardial Enhancement in Pediatric Hypertrophic Cardiomyopathy: Correlation with LV Function, Echocardiography, and Demographic Parameters. Pediatr. Cardiol. 2017, 38, 1024–1031. [Google Scholar] [CrossRef]
- Marian, A.J.; Roberts, R. The molecular genetic basis for hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2001, 33, 655–670. [Google Scholar] [CrossRef] [Green Version]
- Nagueh, S.F.; McFalls, J.; Meyer, D.; Hill, R.; Zoghbi, W.A.; Tam, J.W.; Quinones, M.A.; Roberts, R.; Marian, A.J. Tissue Doppler imaging predicts the development of hypertrophic cardiomyopathy in subjects with subclinical disease. Circulation 2003, 108, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maskatia, S.A.; Decker, J.A.; Spinner, J.A.; Kim, J.J.; Price, J.F.; Jefferies, J.L.; Dreyer, W.J.; Smith, E.O.; Rossano, J.W.; Denfield, S.W. Restrictive physiology is associated with poor outcomes in children with hypertrophic cardiomyopathy. Pediatr. Cardiol. 2012, 33, 141–149. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.J.; Nagueh, S.F.; Pignatelli, R.H.; Denfield, S.W.; Dreyer, W.J.; Price, J.F.; Clunie, S.; Bezold, L.I.; Hays, A.L.; Towbin, J.A.; et al. Characterization of left ventricular diastolic function by tissue Doppler imaging and clinical status in children with hypertrophic cardiomyopathy. Circulation 2004, 109, 1756–1762. [Google Scholar] [CrossRef] [Green Version]
- Kaski, J.P.; Tome-Esteban, M.T.; Mead-Regan, S.; Pantazis, A.; Marek, J.; Deanfield, J.E.; McKenna, W.J.; Elliott, P.M. B-type natriuretic peptide predicts disease severity in children with hypertrophic cardiomyopathy. Heart 2008, 94, 1307–1311. [Google Scholar] [CrossRef]
- Minami, Y.; Haruki, S.; Kanbayashi, K.; Maeda, R.; Itani, R.; Hagiwara, N. B-type natriuretic peptide and risk of sudden death in patients with hypertrophic cardiomyopathy. Heart Rhythm. 2018, 15, 1484–1490. [Google Scholar] [CrossRef]
- Ziółkowska, L.; Mazurkiewicz, Ł.; Petryka, J.; Kowalczyk-Domagała, M.; Boruc, A.; Bieganowska, K.; Ciara, E.; Piekutowska-Abramczuk, D.; Śpiewak, M.; Miśko, J.; et al. The Indices of Cardiovascular Magnetic Resonance Derived Atrial Dynamics May Improve the Contemporary Risk Stratification Algorithms in Children with Hypertrophic Cardiomyopathy. J. Clin. Med. 2021, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Ganame, J.; Mertens, L.; Eidem, B.W.; Claus, P.; D’Hooge, J.; Havemann, L.M.; McMahon, C.J.; Elayda, M.A.; Vaughn, W.K.; Towbin, J.A.; et al. Regional myocardial deformation in children with hypertrophic cardiomyopathy: Morphological and clinical correlations. Eur. Heart J. 2007, 28, 2886–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, C.J.; Ackerman, M.J.; Nishimura, R.A.; Tajik, A.J.; Gersh, B.J.; Ommen, S.R. Outcome of patients with hypertrophic cardiomyopathy and a normal electrocardiogram. J. Am. Coll. Cardiol. 2009, 54, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayosi, B.M.; Keavney, B.; Kardos, A.; Davies, C.H.; Ratcliffe, P.J.; Farrall, M.; Watkins, H. Electrocardiographic measures of left ventricular hypertrophy show greater heritability than echocardiographic left ventricular mass. Eur. Heart J. 2002, 23, 1963–1971. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Nelson, C.P.; Gaunt, T.R.; van der Harst, P.; Barnes, T.; Braund, P.S.; Lawlor, D.A.; Casas, J.P.; Padmanabhan, S.; Drenos, F.; et al. Four genetic loci influencing electrocardiographic indices of left ventricular hypertrophy. Circ. Cardiovasc. Genet. 2011, 4, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Haghjoo, M.; Mohammadzadeh, S.; Taherpour, M.; Faghfurian, B.; Fazelifar, A.F.; Alizadeh, A.; Rad, M.A.; Sadr-Ameli, M.A. ST-segment depression as a risk factor in hypertrophic cardiomyopathy. Europace 2009, 11, 643–649. [Google Scholar] [CrossRef]
- Östman-Smith, I.; Wisten, A.; Nylander, E.; Bratt, E.L.; de-Wahl Granelli, A.; Oulhaj, A.; Ljungström, E. Electrocardiographic amplitudes: A new risk factor for sudden death in hypertrophic cardiomyopathy. Eur. Heart J. 2010, 31, 439–449. [Google Scholar] [CrossRef]
- Norrish, G.; Topriceanu, C.; Qu, C.; Field, E.; Walsh, H.; Ziółkowska, L.; Olivotto, I.; Passantino, S.; Favilli, S.; Anastasakis, A.; et al. The role of the electrocardiographic phenotype in risk stratification for sudden cardiac death in childhood hypertrophic cardiomyopathy. Eur. J. Prev. Cardiol. 2021, 29, 645–653. [Google Scholar] [CrossRef]
- Cortez, D.; Graw, S.; Mestroni, L. In Hypertrophic Cardiomyopathy, the Spatial Peaks QRS-T Angle Identifies Those With Sustained Ventricular Arrhythmias. Clin. Cardiol. 2016, 39, 459–463. [Google Scholar] [CrossRef]
- Gray, B.; Ingles, J.; Medi, C.; Semsarian, C. Prolongation of the QTc interval predicts appropriate implantable cardioverter-defibrillator therapies in hypertrophic cardiomyopathy. JACC Heart Fail. 2013, 1, 149–155. [Google Scholar] [CrossRef]
- Debonnaire, P.; Katsanos, S.; Joyce, E.; Van den Brink, O.V.; Atsma, D.E.; Schalij, M.J.; Bax, J.J.; Delgado, V.; Marsan, N.A. QRS Fragmentation and QTc Duration Relate to Malignant Ventricular Tachyarrhythmias and Sudden Cardiac Death in Patients with Hypertrophic Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2015, 26, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Molloy, T.J.; Okin, P.M.; Devereux, R.B.; Kligfield, P. Electrocardiographic detection of left ventricular hypertrophy by the simple QRS voltage-duration product. J. Am. Coll. Cardiol. 1992, 20, 1180–1186. [Google Scholar] [CrossRef] [Green Version]
- Walinder Osterberg, A.; Beausejour-Ladouceur, V.; Stephenson, E.A.; Mital, S. Independent validation of the ECG Risk Score for Sudden Death Risk stratification in Pediatric Hypertrophic Cardiomyopathy. Circulation 2021, 144, A10353. [Google Scholar] [CrossRef]
- Wålinder Österberg, A.; Östman-Smith, I.; Jablonowski, R.; Carlsson, M.B.; Green, H.; Gunnarsson, C.; Liuba, P.; Fernlund, E. High ECG risk-scores predict late gadolinium enhancement on magnetic resonance imaging in HCM in the young. Pediatr. Cardiol. 2020, 43, 492–500. [Google Scholar]
- Östman-Smith, I.; Sjöberg, G.; Larsson, P.; Rydberg, A.; Fernlund, E. Risk factors for sudden death in childhood—Differencies and similarities between non-syndrome associated hypertrophic cardiomyopathy and Noonan-group syndrome associated hypertrophic cardiomyopathy. Cardiol. Young. 2021, 31. in press. [Google Scholar]
- Kautzner, J.; Yi, G.; Camm, A.J.; Malik, M. Short- and long-term reproducibility of QT, QTc, and QT dispersion measurement in healthy subjects. Pacing Clin. Electrophysiol. 1994, 17, 928–937. [Google Scholar] [CrossRef]
- Crilley, J.G.; Boehm, E.A.; Blair, E.; Rajagopalan, B.; Blamire, A.M.; Styles, P.; McKenna, W.J.; Ostman-Smith, I.; Clarke, K.; Watkins, H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J. Am. Coll. Cardiol. 2003, 41, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Raman, B.; Ariga, R.; Spartera, M.; Sivalokanathan, S.; Chan, K.; Dass, S.; Petersen, S.E.; Daniels, M.J.; Francis, J.; Smillie, R.; et al. Progression of myocardial fibrosis in hypertrophic cardiomyopathy: Mechanisms and clinical implications. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Jablonowski, R.; Fernlund, E.; Aletras, A.H.; Engblom, H.; Heiberg, E.; Liuba, P.; Arheden, H.; Carlsson, M. Regional Stress-Induced Ischemia in Non-fibrotic Hypertrophied Myocardium in Young HCM Patients. Pediatr. Cardiol. 2015, 36, 1662–1669. [Google Scholar] [CrossRef] [Green Version]
- Raphael, C.E.; Cooper, R.; Parker, K.H.; Collinson, J.; Vassiliou, V.; Pennell, D.J.; de Silva, R.; Hsu, L.Y.; Greve, A.M.; Nijjer, S.; et al. Mechanisms of Myocardial Ischemia in Hypertrophic Cardiomyopathy: Insights from Wave Intensity Analysis and Magnetic Resonance. J. Am. Coll. Cardiol. 2016, 68, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Yetman, A.T.; McCrindle, B.W.; MacDonald, C.; Freedom, R.M.; Gow, R. Myocardial bridging in children with hypertrophic cardiomyopathy—A risk factor for sudden death. N. Engl. J. Med. 1998, 339, 1201–1209. [Google Scholar] [CrossRef]
- Biagini, E.; Pazzi, C.; Olivotto, I.; Musumeci, B.; Limongelli, G.; Boriani, G.; Pacileo, G.; Mastromarino, V.; Bacchi Reggiani, M.L.; Lorenzini, M.; et al. Usefulness of Electrocardiographic Patterns at Presentation to Predict Long-term Risk of Cardiac Death in Patients With Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2016, 118, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, A.P.; Stamatopoulos, I.; Antoniades, C.; Anastasakis, A.; Kotsiopoulou, C.; Theopistou, A.; Misailidou, M.; Fourlas, C.; Elliott, P.M.; Stefanadis, C. ST segment “hump” during exercise testing and the risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Ann. Noninvasive Electrocardiol. 2009, 14, 158–164. [Google Scholar] [CrossRef]
- Spinner, J.A.; Noel, C.V.; Denfield, S.W.; Krishnamurthy, R.; Jeewa, A.; Dreyer, W.J.; Maskatia, S.A. Association of Late Gadolinium Enhancement and Degree of Left Ventricular Hypertrophy Assessed on Cardiac Magnetic Resonance Imaging With Ventricular Tachycardia in Children With Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2016, 117, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Axelsson Raja, A.; Farhad, H.; Valente, A.M.; Couce, J.P.; Jefferies, J.L.; Bundgaard, H.; Zahka, K.; Lever, H.; Murphy, A.M.; Ashley, E.; et al. Prevalence and Progression of Late Gadolinium Enhancement in Children and Adolescents with Hypertrophic Cardiomyopathy. Circulation 2018, 138, 782–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonura, E.D.; Bos, J.M.; Abdelsalam, M.A.; Araoz, P.A.; Ommen, S.R.; Ackerman, M.J.; Geske, J.B. Cardiac Magnetic Resonance Imaging Features in Hypertrophic Cardiomyopathy Diagnosed at <21 Years of Age. Am. J. Cardiol. 2020, 125, 1249–1255. [Google Scholar] [CrossRef]
- Chaowu, Y.; Shihua, Z.; Jian, L.; Li, L.; Wei, F. Cardiovascular magnetic resonance characteristics in children with hypertrophic cardiomyopathy. Circ. Heart Fail. 2013, 6, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Petryka-Mazurkiewicz, J.; Ziolkowska, L.; Kowalczyk-Domagala, M.; Mazurkiewicz, L.; Boruc, A.; Spiewak, M.; Misko, J.; Bieganowska, K.; Marczak, M.; Brzezinska-Rajszys, G. LGE for Risk Stratification in Primary Prevention in Children with HCM. JACC Cardiovasc. Imaging 2020, 13, 2684–2686. [Google Scholar] [CrossRef]
- Norrish, G.; Qu, C.; Field, E.; Cervi, E.; Khraiche, D.; Klaassen, S.; Ojala, T.H.; Sinagra, G.; Yamazawa, H.; Marrone, C.; et al. External validation of the HCM Risk-Kids model for predicting sudden cardiac death in childhood hypertrophic cardiomyopathy. Eur. J. Prev. Cardiol. 2021, 29, 678–686. [Google Scholar] [CrossRef]
- Fernlund, E.; Sjöberg, G.; Alenius Dahlqvist, J.; Larsson, P.; Östman-Smith, I. Assessment in a geographical cohort of PRIMaCY performance for risk stratification for sudden cardiac death in childhood hypertrophic cardiomyopathy. Cardiol. Young 2022, 32. in press. [Google Scholar]
- McKenna, W.J.; Stewart, J.T.; Nihoyannopoulos, P.; McGinty, F.; Davies, M.J. Hypertrophic cardiomyopathy without hypertrophy: Two families with myocardial disarray in the absence of increased myocardial mass. Br. Heart J. 1990, 63, 287–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnava, A.; Baboonian, C.; Davison, F.; de Cruz, L.; Elliott, P.M.; Davies, M.J.; McKenna, W.J. A new mutation of the cardiac troponin T gene causing familial hypertrophic cardiomyopathy without left ventricular hypertrophy. Heart 1999, 82, 621–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppini, R.; Ho, C.Y.; Ashley, E.; Day, S.; Ferrantini, C.; Girolami, F.; Tomberli, B.; Bardi, S.; Torricelli, F.; Cecchi, F.; et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J. Am. Coll. Cardiol. 2014, 64, 2589–2600. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, G.; Pacileo, G.; Calabrò, R. Is sudden cardiac death predictable in LEOPARD syndrome? Cardiol. Young. 2006, 16, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, C.; Voges, I.; Daubeney, P.E.F. Out-of-hospital cardiac arrest and survival in a patient with Noonan syndrome and multiple lentigines: A case report. J. Med. Case Rep. 2019, 13, 194. [Google Scholar] [CrossRef]
- Jorholt, J.; Formicheva, Y.; Vershinina, T.; Kiselev, A.; Muravyev, A.; Demchenko, E.; Fedotov, P.; Zlotina, A.; Rygkov, A.; Vasichkina, E.; et al. Two New Cases of Hypertrophic Cardiomyopathy and Skeletal Muscle Features Associated with ALPK3 Homozygous and Compound Heterozygous Variants. Genes 2020, 11, 1201. [Google Scholar] [CrossRef]
- Wolf, C.M.; Zenker, M.; Burkitt-Wright, E.; Edouard, T.; García-Miñaúr, S.; Lebl, J.; Shaikh, G.; Tartaglia, M.; Verloes, A.; Östman-Smith, I. Management of cardiac aspects in children with Noonan syndrome—Results from a European clinical practice survey among paediatric cardiologists. Eur. J. Med. Genet. 2022, 65, 104372. [Google Scholar] [CrossRef]
- Sherrid, M.V. Implantable cardioverter-defibrillators for children and adolescents at high risk for sudden death from hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2013, 61, 1536–1538. [Google Scholar] [CrossRef]
- Christiaans, I.; van Engelen, K.; van Langen, I.M.; Birnie, E.; Bonsel, G.J.; Elliott, P.M.; Wilde, A.A. Risk stratification for sudden cardiac death in hypertrophic cardiomyopathy: Systematic review of clinical risk markers. Europace 2010, 12, 313–321. [Google Scholar] [CrossRef]





| Morphological Characteristics | Ped HCM Refs | Adult HCM Refs | ||
|---|---|---|---|---|
| Points | ||||
| Deviation of QRS-axis present | Yes | 1 | [19] | [58] |
| Pathological T-wave-inversion ≥1 mm | [19] | [58] | ||
| present In limb-lead | Yes | 1 | [19] | [58] |
| In precordial lead | Yes | 2 | ||
| In both limb-lead and precordial lead | Yes | 2 | [19,59] | [58] |
| ST-segment depression ≥ 2 mm present | Yes | 2 | [19] | [57,58] |
| S-wave greater than R-wave in lead V4 | Yes | 2 | [19] | [58] |
| ECG measurements | ||||
| Six limb-lead QRS-amplitude sum in mV | 0–7.6 mV ≥7.7–9.9 mV ≥10.0–11.9 mV ≥12.0 mV | 0 1 2 3 | [9,12,19] | [58] |
| 12-lead QRS-amplitude x duration product mV.sec | 0–2.19 mV.sec ≥2.2–2.49 mV.sec ≥2.5–2.99 mV.sec ≥3.0 mV.sec | 0 1 2 3 | [19] | [58] |
| QTc (Bazetts formula) | <440 msec | 0 | ||
| ≥440 msec | 1 | [19,25,60] | [58,61,62] | |
| Maximal total score | 14 points | |||
| Parameter | C-Statistic [95%CI] | Sens | Spec | PPV | NPV | %TP | Mean FU | Age at D | End -Pts | Patient Years |
|---|---|---|---|---|---|---|---|---|---|---|
| Side-by-side comparison in the same cohort [29] | ||||||||||
| ESC ≥ 2RF | 0.66 [0.47–0.85] | 45 | 86 | 28 | 93 | 17 | 13.4 | 10.9 | 11 | 1474 [29] |
| AHA ≥ 1RF | 0.55 [0.37–0.73] | 55 | 56 | 13 | 91 | 45 | ||||
| Max wth Det.Z-sc ≥ 4.5 | 0.79 [0.66–0.92] | 90 | 68 | 24 | 98 | 38 | ||||
| ECG Risc ≥ 6 | 0.87 [0.80–0.94] | 100 | 73 | 31 | 100 | 40 | ||||
| HCMRiskKids ≥ 6 (ext validation 1) | 0.69 [0.64–0.89] | 73 | 65 | 22 | 95 | 39 | ||||
| HRK+ECGri ≥ 14 | 0.82 [0.68–0.96] | 82 | 82 | 38 | 97 | 26 | ||||
| 7plHRK+ECGri ≥ 14 | 0.90 [0.83–0.96] | 100 | 77 | 38 | 100 | 32 | ||||
| Studies with evaluation of single parameter cut-offs | ||||||||||
| HCMRiskKids ≥ 6% (compl values cohort) | 0.69 [0.66–0.72] | 76 | 58 | 12 | 97 | 44 | 5.3 | 11 | 34 | 3005 [28] |
| HCMRiskKids ≥ 6% (ext.validation 2) | 0.70 [0.60–0.81] | 74 | 73 | 14 | 98 | 30 | 3.5 | 12.3 | 23 | 1474 [80] |
| PRIMaCY ≥ 9% (ext validation 2) | 0.66 [0.49–0.84] | 82 | 52 | 20 | 95 | 40 | 10.6 | 10.6 | 11 | 1463 [81] |
| ECG Risc ≥ 6 (external validation) | 0.76 [?] | 95 | 56 | 28 | 99 | 47 | 14.6 | ? | 22 | 2102 [64] |
| Studies evaluation parameters as continuous functions | ||||||||||
| Max wthDet Z-score | 0.79 [0.65–0.92] | 13.4 | 10.9 | 11 | 1474 [29] | |||||
| ECG risk score | 0.91 [0.85–0.97] | 13.4 | 10.9 | 11 | 1474 [29] | |||||
| HCMRisk-Kids (ext validation 1) | 0.77 [0.62–0.93] | 13.4 | 10.9 | 11 | 1474 [29] | |||||
| HCMRisk-Kids (ext validation 2) | 0.75 [0.52–0.97] | 3.5 | 12.3 | 23 | 1474 [80] | |||||
| PRIMaCY (derivation cohort) | 0.75 [?] | 5.0 | 9.8 | 53 | 2855 [18] | |||||
| PRIMaCY (ext validation cohort1) | 0.71 [?] | 4.9 | 13.8 | 22 | 1400 [18] | |||||
| PRIMaCY (external validation 2) | 0.71 [0.51–0.90] | 10.6 | 10.6 | 11 | 1463 [81] | |||||
| Parameter | Age at Diagnosis (yrs) | Age at SCD (yrs) | Detroit Z-Score (M-mode) | Boston Z-Score (2-D) | Max Wth (mm) | Post LV Wall Detroit Z-Score n = 7 | QTc (ms) n = 12 | Limb-Lead QRS-Sum (mV) n = 8 | ECG Risk Score n = 8 |
|---|---|---|---|---|---|---|---|---|---|
| Median | 7.2 | 13.2 | 6.8 | 27.4 | 28 | 6.1 | 478 | 17.0 | 11 |
| IQR | 0.2–11.5 | 9.9–26 | 6.1–7.8 | 20.8–36 | 24–33 | 4.9–6.8 | 439–500 | 11.4–37 | 8–11 |
| Range | 0.01–17 | 2.0–42 | 4.7–8.7 | 13.3–43 | 17–40 | 2.7–8.9 | 408–528 | 10.9–42 | 7–13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Östman-Smith, I. What Aspects of Phenotype Determine Risk for Sudden Cardiac Death in Pediatric Hypertrophic Cardiomyopathy? J. Cardiovasc. Dev. Dis. 2022, 9, 124. https://doi.org/10.3390/jcdd9050124
Östman-Smith I. What Aspects of Phenotype Determine Risk for Sudden Cardiac Death in Pediatric Hypertrophic Cardiomyopathy? Journal of Cardiovascular Development and Disease. 2022; 9(5):124. https://doi.org/10.3390/jcdd9050124
Chicago/Turabian StyleÖstman-Smith, Ingegerd. 2022. "What Aspects of Phenotype Determine Risk for Sudden Cardiac Death in Pediatric Hypertrophic Cardiomyopathy?" Journal of Cardiovascular Development and Disease 9, no. 5: 124. https://doi.org/10.3390/jcdd9050124