
Molecular Mechanism Underlying Role of the XBP1s in Cardiovascular Diseases
Abstract
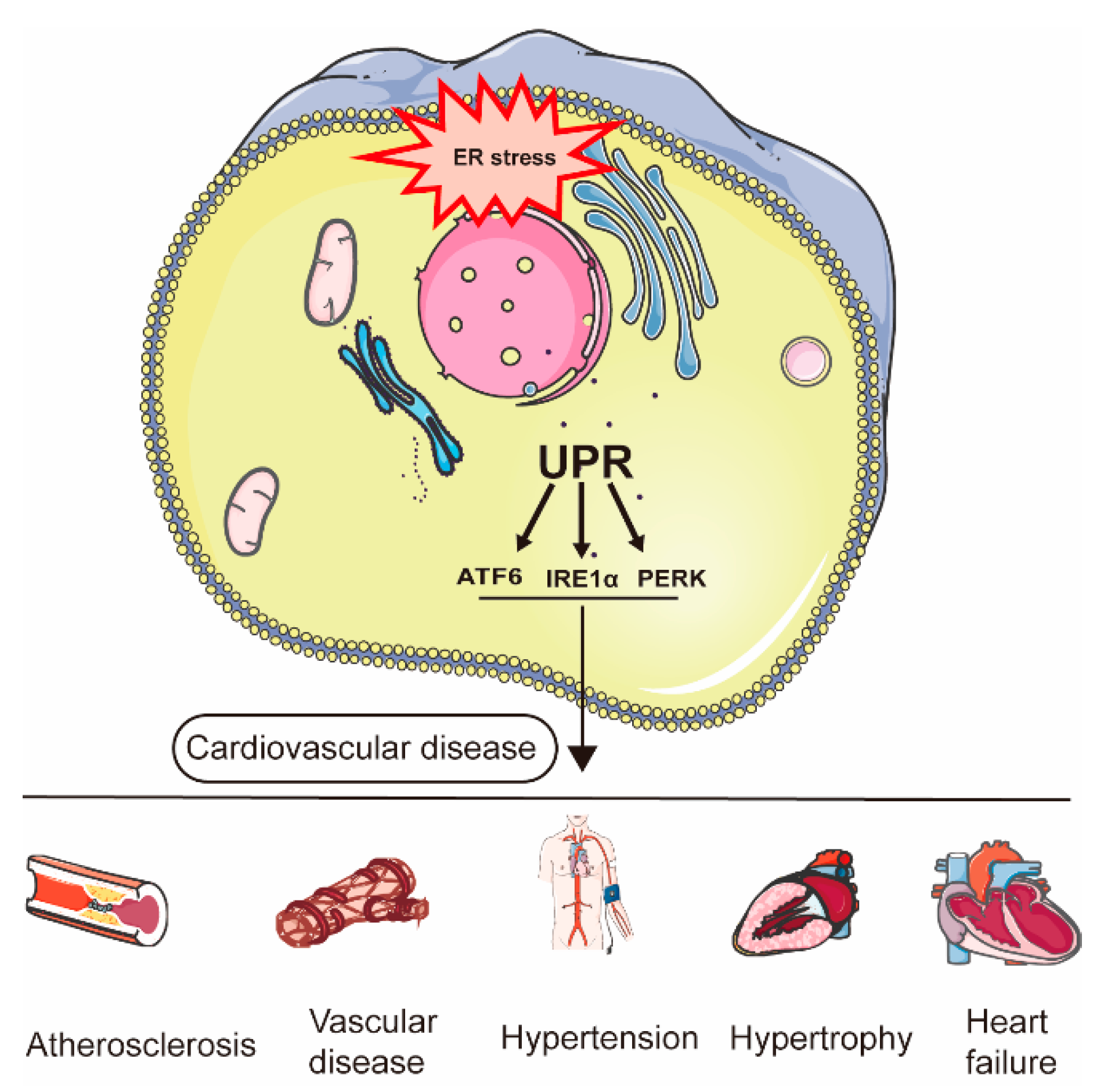
:1. Introduction
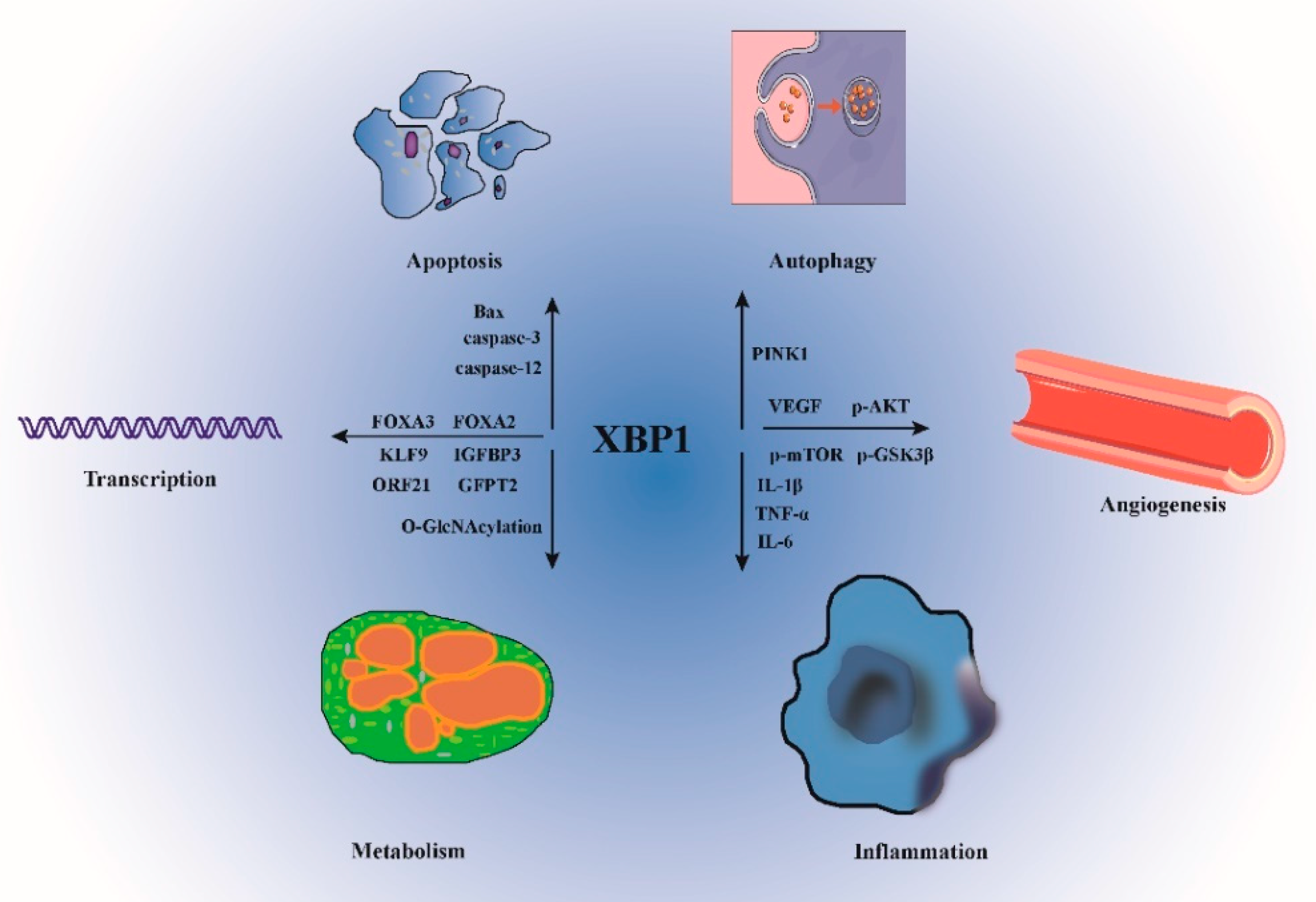
2. XBP1s Structure and Cellular Function
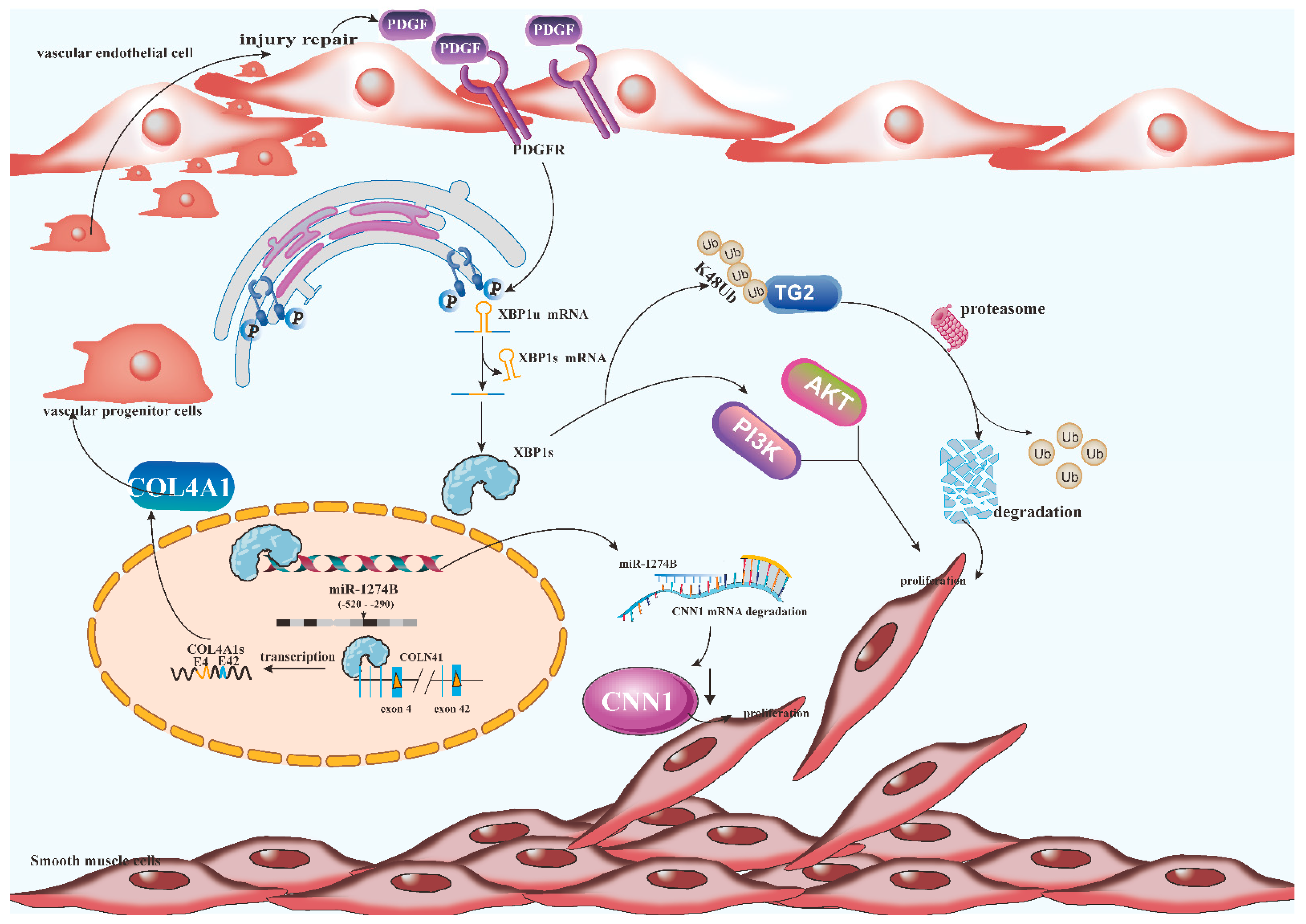
3. The Role of XBP1s in Smooth Muscle Cells
4. The Role of XBP1s in Ischemia/Reperfusion Injury and Atherosclerosis
5. The Role of XBP1s in Hypertension
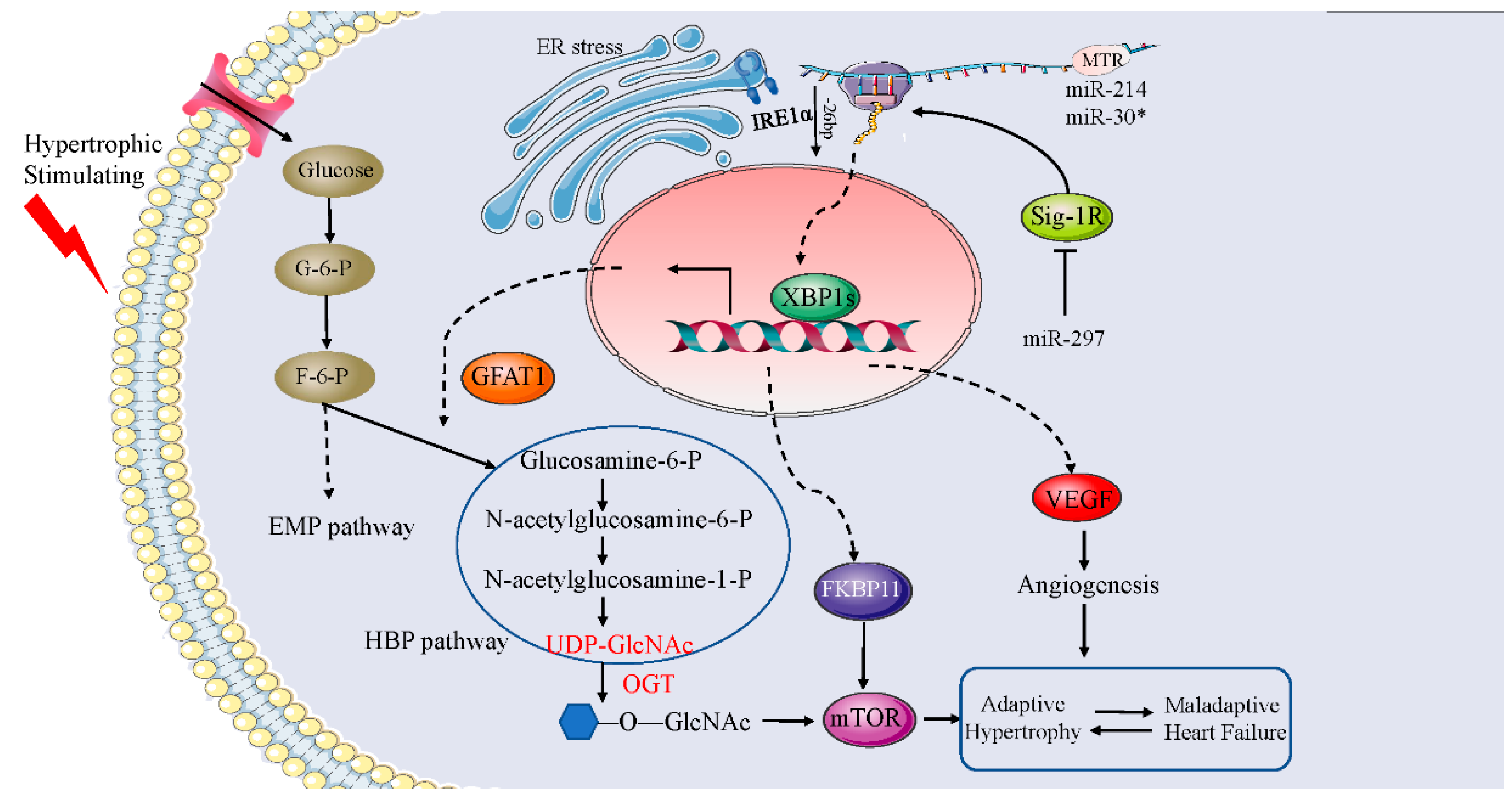
6. The Role of XBP1s in Cardiac Hypertrophy
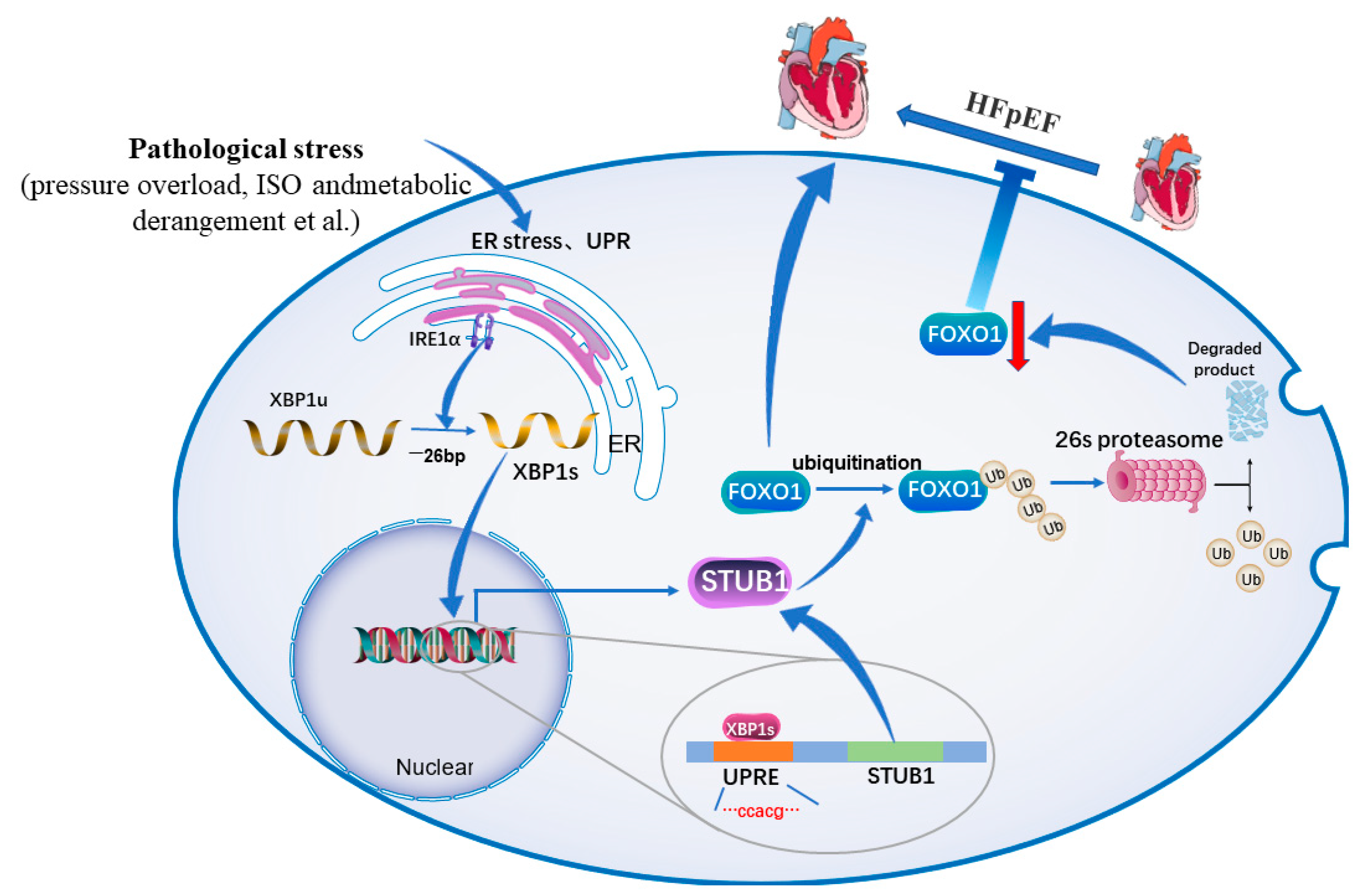
7. The Role of XBP1s in Heart Failure
8. Therapeutic Potential of XBP1s in Cardiovascular Diseases
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| XBP1s | Spliced X-box binding protein-1 |
| CVDs | Cardiovascular diseases |
| ERS | Endoplasmic reticulum stress |
| UPR | Unfolded protein responses |
| ERAD | ER-associated degradation |
| IRE1a | Inositol-requiring kinase 1 |
| ATF6 | Activating transcription factor 6 |
| PERK | Protein kinase-like ER kinase |
| eIF2α | Eukaryotic translation initiation factor 2α |
| ATF4 | Activating transcription factor 4 |
| CREB | cAMP-response element-binding |
| bZIP | Basic-region leucine zipper |
| SMCs | Smooth muscle cells |
| VEGF | Vascular endothelial cell growth factor |
| PDGF | Platelet-derived growth factor |
| bFGF | Basic fibroblast growth factor |
| CNN1 | Calponin h1 |
| COL4A1 | Collagen alpha 1 |
| VPCs | Vascular progenitor cells |
| TG2 | Transglutaminase 2 |
| TLR | Toll-like receptors |
| ROS | Reactive oxygen species |
| RAAS | Renin-angiotensin-aldosterone system |
| HBP | Hexosamine biosynthetic pathway |
| Gfat1 | Glutamine: fructose-6-phosphate amidotransferase 1 |
| mTOR | Mechanistic target of rapamycin |
| FKBP11 | FK506-binding protein 11 |
| NOX4 | NADPH oxidase 4 |
| Sig-1R | Sigma-1receptor |
| HFpEF | Heart failure with preserved ejection fraction |
| HFmrEF | Heart failure with mildly reduced ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| LVEF | Left ventricular ejection fraction |
| BNP | Brain natriuretic peptide |
| FOXO1 | Forkhead box protein O1 |
| STUB1 | STIP1 homology and U-Box-containing protein 1 |
References
- Meier, T.; Gräfe, K.; Senn, F.; Sur, P.; Stangl, G.I.; Dawczynski, C.; März, W.; Kleber, M.E.; Lorkowski, S. Cardiovascular mortality attributable to dietary risk factors in 51 countries in the WHO European Region from 1990 to 2016: A systematic analysis of the Global Burden of Disease Study. Eur. J. Epidemiol. 2019, 34, 37–55. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. CMLS 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Okada, T.; Yoshida, H.; Akazawa, R.; Negishi, M.; Mori, K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366, 585–594. [Google Scholar] [CrossRef]
- Wang, P.; Li, J.; Sha, B. The ER stress sensor PERK luminal domain functions as a molecular chaperone to interact with misfolded proteins. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Chaveroux, C.; Carraro, V.; Canaple, L.; Averous, J.; Maurin, A.C.; Jousse, C.; Muranishi, Y.; Parry, L.; Mesclon, F.; Gatti, E.; et al. In vivo imaging of the spatiotemporal activity of the eIF2α-ATF4 signaling pathway: Insights into stress and related disorders. Sci. Signal. 2015, 8, rs5. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Chen, J.; Hua, X.; Sun, Y.; Cui, R.; Sha, J.; Zhu, X. The emerging role of XBP1 in cancer. Biomed. Pharm. 2020, 127, 110069. [Google Scholar] [CrossRef]
- Yoshida, H. Unconventional splicing of XBP-1 mRNA in the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2323–2333. [Google Scholar] [CrossRef]
- Chang, M.; Liu, G.; Wang, Y.; Lv, H.; Jin, Y. Long non-coding RNA LINC00299 knockdown inhibits ox-LDL-induced T/G HA-VSMC injury by regulating miR-135a-5p/XBP1 axis in atherosclerosis. Panminerva Med. 2022, 64, 38–47. [Google Scholar] [CrossRef]
- Bi, X.; Zhang, G.; Wang, X.; Nguyen, C.; May, H.I.; Li, X.; Al-Hashimi, A.A.; Austin, R.C.; Gillette, T.G.; Fu, G.; et al. Endoplasmic Reticulum Chaperone GRP78 Protects Heart From Ischemia/Reperfusion Injury Through Akt Activation. Circ. Res. 2018, 122, 1545–1554. [Google Scholar] [CrossRef]
- Duan, Q.; Ni, L.; Wang, P.; Chen, C.; Yang, L.; Ma, B.; Gong, W.; Cai, Z.; Zou, M.H.; Wang, D.W. Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell 2016, 15, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Sage, A.P.; Nus, M.; Bagchi Chakraborty, J.; Tsiantoulas, D.; Newland, S.A.; Finigan, A.J.; Masters, L.; Binder, C.J.; Mallat, Z. X-Box Binding Protein-1 Dependent Plasma Cell Responses Limit the Development of Atherosclerosis. Circ. Res. 2017, 121, 270–281. [Google Scholar] [CrossRef]
- Fink, E.E.; Moparthy, S.; Bagati, A.; Bianchi-Smiraglia, A.; Lipchick, B.C.; Wolff, D.W.; Roll, M.V.; Wang, J.; Liu, S.; Bakin, A.V.; et al. XBP1-KLF9 Axis Acts as a Molecular Rheostat to Control the Transition from Adaptive to Cytotoxic Unfolded Protein Response. Cell Rep. 2018, 25, 212–223.e214. [Google Scholar] [CrossRef] [Green Version]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef]
- Luo, Q.; Shi, W.; Dou, B.; Wang, J.; Peng, W.; Liu, X.; Zhao, D.; Tang, F.; Wu, Y.; Li, X.; et al. XBP1- IGFBP3 Signaling Pathway Promotes NSCLC Invasion and Metastasis. Front. Oncol. 2021, 11, 654995. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, B.; Meng, M.; Zhao, W.; Wang, D.; Yuan, Y.; Zheng, Y.; Qiu, J.; Li, Y.; Li, G.; et al. FOXA3 induction under endoplasmic reticulum stress contributes to non-alcoholic fatty liver disease. J. Hepatol. 2021, 75, 150–162. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Yuan, F.; Liu, J.; Li, D.; Wei, L.; Wang, X.; Yuan, L. X-box-binding protein 1 is required for pancreatic development in Xenopus laevis. Acta Biochim. Et Biophys. Sin. 2020, 52, 1215–1226. [Google Scholar] [CrossRef]
- Wang, V.; Davis, D.A.; Deleage, C.; Brands, C.; Choi, H.S.; Haque, M.; Yarchoan, R. Induction of Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Thymidine Kinase (ORF21) by X-Box Binding Protein 1. J. Virol. 2020, 94, e01555-19. [Google Scholar] [CrossRef]
- Prasad, V.; Suomalainen, M.; Jasiqi, Y.; Hemmi, S.; Hearing, P.; Hosie, L.; Burgert, H.G.; Greber, U.F. The UPR sensor IRE1α and the adenovirus E3-19K glycoprotein sustain persistent and lytic infections. Nat. Commun. 2020, 11, 1997. [Google Scholar] [CrossRef] [Green Version]
- Qiao, D.; Skibba, M.; Xu, X.; Garofalo, R.P.; Zhao, Y.; Brasier, A.R. Paramyxovirus replication induces the hexosamine biosynthetic pathway and mesenchymal transition via the IRE1α-XBP1s arm of the unfolded protein response. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2021, 321, L576–L594. [Google Scholar] [CrossRef]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Wang, Z.V.; Tao, C.; Gao, N.; Holland, W.L.; Ferdous, A.; Repa, J.J.; Liang, G.; Ye, J.; Lehrman, M.A.; et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J. Clin. Investig. 2013, 123, 455–468. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Zhu, L.; Du, S.; Mao, J.; Wang, Y.; Wang, S.; Bo, Q.; Tu, Y.; Yi, Q. The P300/XBP1s/Herpud1 axis promotes macrophage M2 polarization and the development of choroidal neovascularization. J. Cell Mol. Med. 2021, 25, 6709–6720. [Google Scholar] [CrossRef]
- Sharmin, M.M.; Hayashi, S.; Miyaji, M.; Ishizaki, H.; Matsuyama, H.; Haga, S.; Yonekura, S. Insulin-like growth factor-1 induces IRE1-XBP1-dependent endoplasmic reticulum biogenesis in bovine mammary epithelial cells. J. Dairy Sci. 2021, 104, 12094–12104. [Google Scholar] [CrossRef]
- Mo, Z.T.; Zheng, J.; Liao, Y.L. Icariin inhibits the expression of IL-1β, IL-6 and TNF-α induced by OGD/R through the IRE1/XBP1s pathway in microglia. Pharm. Biol. 2021, 59, 1473–1479. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q.; Pang, Y.; Xu, X.; Cui, K.; Zhang, Y.; Mai, K.; Ai, Q. Molecular cloning and the involvement of IRE1α-XBP1s signaling pathway in palmitic acid induced—Inflammation in primary hepatocytes from large yellow croaker (Larimichthys crocea). Fish. Shellfish Immunol. 2020, 98, 112–121. [Google Scholar] [CrossRef]
- Luo, R.; Xiao, F.; Wang, P.; Hu, Y.X. lncRNA H19 sponging miR-93 to regulate inflammation in retinal epithelial cells under hyperglycemia via XBP1s. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2020, 69, 255–265. [Google Scholar] [CrossRef]
- Dong, L.; Tan, C.W.; Feng, P.J.; Liu, F.B.; Liu, D.X.; Zhou, J.J.; Chen, Y.; Yang, X.X.; Zhu, Y.H.; Zhu, Z.Q. Activation of TREM-1 induces endoplasmic reticulum stress through IRE-1α/XBP-1s pathway in murine macrophages. Mol. Immunol. 2021, 135, 294–303. [Google Scholar] [CrossRef]
- Rodrigo, S.; Panadero, M.I.; Fauste, E.; Rodríguez, L.; Roglans, N.; Álvarez-Millán, J.J.; Otero, P.; Laguna, J.C.; Bocos, C. Effects of Maternal Fructose Intake on Perinatal ER-Stress: A Defective XBP1s Nuclear Translocation Affects the ER-stress Resolution. Nutrients 2019, 11, 1935. [Google Scholar] [CrossRef] [Green Version]
- El Manaa, W.; Duplan, E.; Goiran, T.; Lauritzen, I.; Vaillant Beuchot, L.; Lacas-Gervais, S.; Morais, V.A.; You, H.; Qi, L.; Salazar, M.; et al. Transcription- and phosphorylation-dependent control of a functional interplay between XBP1s and PINK1 governs mitophagy and potentially impacts Parkinson disease pathophysiology. Autophagy 2021, 17, 4363–4385. [Google Scholar] [CrossRef]
- Woodworth-Hobbs, M.E.; Perry, B.D.; Rahnert, J.A.; Hudson, M.B.; Zheng, B.; Russ Price, S. Docosahexaenoic acid counteracts palmitate-induced endoplasmic reticulum stress in C2C12 myotubes: Impact on muscle atrophy. Physiol. Rep. 2017, 5, e13530. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; Tong, D.; Ferdous, A.; Piristine, H.; Dasgupta, S.; Wang, X.; French, K.M.; Villalobos, E.; et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat. Commun. 2021, 12, 1684. [Google Scholar] [CrossRef]
- Li, R.; Shen, Y.; Li, X.; Lu, L.; Wang, Z.; Sheng, H.; Hoffmann, U.; Yang, W. Activation of the XBP1s/O-GlcNAcylation Pathway Improves Functional Outcome After Cardiac Arrest and Resuscitation in Young and Aged Mice. Shock 2021, 56, 755–761. [Google Scholar] [CrossRef]
- Wang, N.; Ma, J.; Ma, Y.; Lu, L.; Ma, C.; Qin, P.; Gao, E.; Zuo, M.; Yang, J.; Yang, L. Electroacupuncture Pretreatment Mitigates Myocardial Ischemia/Reperfusion Injury via XBP1/GRP78/Akt Pathway. Front. Cardiovasc. Med. 2021, 8, 629547. [Google Scholar] [CrossRef]
- Tian, J.H.; Wu, Q.; He, Y.X.; Shen, Q.Y.; Rekep, M.; Zhang, G.P.; Luo, J.D.; Xue, Q.; Liu, Y.H. Zonisamide, an antiepileptic drug, alleviates diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress. Acta Pharmacol. Sin. 2021, 42, 393–403. [Google Scholar] [CrossRef]
- Chang, X.; Tian, M.; Zhang, Q.; Liu, F.; Gao, J.; Li, S.; Liu, H.; Hou, X.; Li, L.; Li, C.; et al. Grape seed proanthocyanidin extract ameliorates cisplatin-induced testicular apoptosis via PI3K/Akt/mTOR and endoplasmic reticulum stress pathways in rats. J. Food Biochem. 2021, 45, e13825. [Google Scholar] [CrossRef]
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. CCS 2020, 18, 12. [Google Scholar] [CrossRef] [Green Version]
- Barez, S.R.; Atar, A.M.; Aghaei, M. Mechanism of inositol-requiring enzyme 1-alpha inhibition in endoplasmic reticulum stress and apoptosis in ovarian cancer cells. J. Cell Commun. Signal. 2020, 14, 403–415. [Google Scholar] [CrossRef]
- Sun, X.; Chen, C.; Liu, H.; Tang, S. High glucose induces HSP47 expression and promotes the secretion of inflammatory factors through the IRE1α/XBP1/HIF-1α pathway in retinal Müller cells. Exp. Ther. Med. 2021, 22, 1411. [Google Scholar] [CrossRef]
- Moszyńska, A.; Collawn, J.F.; Bartoszewski, R. IRE1 Endoribonuclease Activity Modulates Hypoxic HIF-1α Signaling in Human Endothelial Cells. Biomolecules 2020, 10, 895. [Google Scholar] [CrossRef]
- Zhang, M.; Tang, M.; Wu, Q.; Wang, Z.; Chen, Z.; Ding, H.; Hu, X.; Lv, X.; Zhao, S.; Sun, J.; et al. LncRNA DANCR attenuates brain microvascular endothelial cell damage induced by oxygen-glucose deprivation through regulating of miR-33a-5p/XBP1s. Aging 2020, 12, 1778–1791. [Google Scholar] [CrossRef]
- Shi, S.; Tang, M.; Li, H.; Ding, H.; Lu, Y.; Gao, L.; Wu, Q.; Zhou, L.; Fu, Y.; Xiao, B.; et al. X-box binding protein l splicing attenuates brain microvascular endothelial cell damage induced by oxygen-glucose deprivation through the activation of phosphoinositide 3-kinase/protein kinase B, extracellular signal-regulated kinases, and hypoxia-inducible factor-1α/vascular endothelial growth factor signaling pathways. J. Cell. Physiol. 2019, 234, 9316–9327. [Google Scholar] [CrossRef]
- Takahashi, N.; Harada, M.; Hirota, Y.; Zhao, L.; Yoshino, O.; Urata, Y.; Izumi, G.; Takamura, M.; Hirata, T.; Koga, K.; et al. A potential role of endoplasmic reticulum stress in development of ovarian hyperstimulation syndrome. Mol. Cell. Endocrinol. 2016, 428, 161–169. [Google Scholar] [CrossRef]
- Kroetsch, J.T.; Levy, A.S.; Zhang, H.; Aschar-Sobbi, R.; Lidington, D.; Offermanns, S.; Nedospasov, S.A.; Backx, P.H.; Heximer, S.P.; Bolz, S.S. Constitutive smooth muscle tumour necrosis factor regulates microvascular myogenic responsiveness and systemic blood pressure. Nat. Commun. 2017, 8, 14805. [Google Scholar] [CrossRef]
- Scott, R.A.; Panitch, A. Decorin mimic regulates platelet-derived growth factor and interferon-γ stimulation of vascular smooth muscle cells. Biomacromolecules 2014, 15, 2090–2103. [Google Scholar] [CrossRef] [Green Version]
- Walker, T.R.; Moore, S.M.; Lawson, M.F.; Panettieri, R.A., Jr.; Chilvers, E.R. Platelet-derived growth factor-BB and thrombin activate phosphoinositide 3-kinase and protein kinase B: Role in mediating airway smooth muscle proliferation. Mol. Pharmacol. 1998, 54, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Kong, L.; Luan, S.; Qi, C.; Wu, F. Ligustrazine Suppresses Platelet-Derived Growth Factor-BB-Induced Pulmonary Artery Smooth Muscle Cell Proliferation and Inflammation by Regulating the PI3K/AKT Signaling Pathway. Am. J. Chin. Med. 2021, 49, 437–459. [Google Scholar] [CrossRef]
- Mao, Y.; Liu, X.Q.; Song, Y.; Zhai, C.G.; Xu, X.L.; Zhang, L.; Zhang, Y. Fibroblast growth factor-2/platelet-derived growth factor enhances atherosclerotic plaque stability. J. Cell Mol. Med. 2020, 24, 1128–1140. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Li, Y.; Yang, J.; Wang, G.; Margariti, A.; Xiao, Q.; Zampetaki, A.; Yin, X.; Mayr, M.; Mori, K.; et al. XBP 1-Deficiency Abrogates Neointimal Lesion of Injured Vessels Via Cross Talk With the PDGF Signaling. Arter. Thromb. Vasc. Biol. 2015, 35, 2134–2144. [Google Scholar] [CrossRef] [Green Version]
- Angbohang, A.; Huang, L.; Li, Y.; Zhao, Y.; Gong, Y.; Fu, Y.; Mao, C.; Morales, J.; Luo, P.; Ehteramyan, M.; et al. X-box binding protein 1-mediated COL4A1s secretion regulates communication between vascular smooth muscle and stem/progenitor cells. J. Biol. Chem. 2021, 296, 100541. [Google Scholar] [CrossRef]
- Chen, H.; Qi, L. SUMO modification regulates the transcriptional activity of XBP1. Biochem. J. 2010, 429, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Xiao, Q.; Chen, M.; Margariti, A.; Martin, D.; Ivetic, A.; Xu, H.; Mason, J.; Wang, W.; Cockerill, G.; et al. Vascular endothelial cell growth-activated XBP1 splicing in endothelial cells is crucial for angiogenesis. Circulation 2013, 127, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.Y.; Minamino, T.; Tsukamoto, O.; Sawada, T.; Asai, M.; Kato, H.; Asano, Y.; Fujita, M.; Takashima, S.; Hori, M.; et al. Overexpression of endoplasmic reticulum-resident chaperone attenuates cardiomyocyte death induced by proteasome inhibition. Cardiovasc. Res. 2008, 79, 600–610. [Google Scholar] [CrossRef]
- Sawada, T.; Minamino, T.; Fu, H.Y.; Asai, M.; Okuda, K.; Isomura, T.; Yamazaki, S.; Asano, Y.; Okada, K.; Tsukamoto, O.; et al. X-box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE-like element in cardiomyocytes. J. Mol. Cell Cardiol. 2010, 48, 1280–1289. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Martinet, W.; Croons, V.; Timmermans, J.P.; Herman, A.G.; De Meyer, G.R. Nitric oxide selectively depletes macrophages in atherosclerotic plaques via induction of endoplasmic reticulum stress. Br. J. Pharm. 2007, 152, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Anagnostopoulou, A.; Camargo, L.L.; Rios, F.J.; Montezano, A.C. Vascular Biology of Superoxide-Generating NADPH Oxidase 5-Implications in Hypertension and Cardiovascular Disease. Antioxid. Redox Signal. 2019, 30, 1027–1040. [Google Scholar] [CrossRef] [Green Version]
- Camargo, L.L.; Harvey, A.P.; Rios, F.J.; Tsiropoulou, S.; Da Silva, R.N.O.; Cao, Z.; Graham, D.; McMaster, C.; Burchmore, R.J.; Hartley, R.C.; et al. Vascular Nox (NADPH Oxidase) Compartmentalization, Protein Hyperoxidation, and Endoplasmic Reticulum Stress Response in Hypertension. Hypertension 2018, 72, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Targeting the Renin-Angiotensin-Aldosterone System to Prevent Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef]
- Duan, Q.; Song, P.; Ding, Y.; Zou, M.H. Activation of AMP-activated protein kinase by metformin ablates angiotensin II-induced endoplasmic reticulum stress and hypertension in mice in vivo. Br. J. Pharm. 2017, 174, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Liu, S.; Wang, H.; Hu, M.; Xing, C.; Song, L. Arsenite Induces Vascular Endothelial Cell Dysfunction by Activating IRE1alpha/XBP1s/HIF1alpha-Dependent ANGII Signaling. Toxicol. Sci. 2017, 160, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhou, M.; Wen, G.; Huang, Y.; Wu, J.; Peng, L.; Jiang, W.; Yuan, H.; Lu, Y.; Cai, J. Paroxetine Attenuates Cardiac Hypertrophy Via Blocking GRK2 and ADRB1 Interaction in Hypertension. J. Am. Heart Assoc. 2021, 10, e016364. [Google Scholar] [CrossRef]
- Ferrara, L.A.; Cardoni, O.; Mancini, M.; Zanchetti, A. Metabolic syndrome and left ventricular hypertrophy in a general population. Results from the Gubbio Study. J. Hum. Hypertens. 2007, 21, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.H.; May, H.I.; Li, Q.; Luo, X.; Huang, J.; Zhang, G.; Niewold, E.; Wang, X.; Gillette, T.G.; Deng, Y.; et al. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat. Commun. 2020, 11, 1771. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Deng, Y.; Zhang, G.; Li, C.; Ding, G.; May, H.I.; Tran, D.H.; Luo, X.; Jiang, D.S.; Li, D.L.; et al. Spliced X-box Binding Protein 1 Stimulates Adaptive Growth Through Activation of mTOR. Circulation 2019, 140, 566–579. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, M.; Li, J.; Wang, Y.; Bao, Q.; Wu, S.; Deng, X.; Tang, X.; Wu, W.; Liu, X. Critical role of X-box binding protein 1 in NADPH oxidase 4-triggered cardiac hypertrophy is mediated by receptor interacting protein kinase 1. Cell Cycle 2017, 16, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Seok, H.; Lee, H.; Lee, S.; Ahn, S.H.; Lee, H.S.; Kim, G.D.; Peak, J.; Park, J.; Cho, Y.K.; Jeong, Y.; et al. Position-specific oxidation of miR-1 encodes cardiac hypertrophy. Nature 2020, 584, 279–285. [Google Scholar] [CrossRef]
- Bao, Q.; Zhao, M.; Chen, L.; Wang, Y.; Wu, S.; Wu, W.; Liu, X. MicroRNA-297 promotes cardiomyocyte hypertrophy via targeting sigma-1 receptor. Life Sci. 2017, 175, 1–10. [Google Scholar] [CrossRef]
- Duan, Q.; Chen, C.; Yang, L.; Li, N.; Gong, W.; Li, S.; Wang, D.W. MicroRNA regulation of unfolded protein response transcription factor XBP1 in the progression of cardiac hypertrophy and heart failure in vivo. J. Transl. Med. 2015, 13, 363. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Sato, M.; Tsumoto, H.; Toba, A.; Soejima, Y.; Arai, T.; Harada, K.; Miura, Y.; Sawabe, M. Proteome analysis demonstrates involvement of endoplasmic reticulum stress response in human myocardium with subclinical left ventricular diastolic dysfunction. Geriatr. Gerontol. Int. 2021, 21, 577–583. [Google Scholar] [CrossRef]
- Park, S.M.; Kang, T.I.; So, J.S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Fu, H.Y.; Sanada, S.; Matsuzaki, T.; Liao, Y.; Okuda, K.; Yamato, M.; Tsuchida, S.; Araki, R.; Asano, Y.; Asanuma, H.; et al. Chemical Endoplasmic Reticulum Chaperone Alleviates Doxorubicin-Induced Cardiac Dysfunction. Circ. Res. 2016, 118, 798–809. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, Z.; Fan, Q.; Guo, J.; Galli, G.; Du, G.; Wang, X.; Xiao, W. Ginkgolide K protects the heart against endoplasmic reticulum stress injury by activating the inositol-requiring enzyme 1α/X box-binding protein-1 pathway. Br. J. Pharm. 2016, 173, 2402–2418. [Google Scholar] [CrossRef] [Green Version]
- Lamb, Y.N. Imeglimin Hydrochloride: First Approval. Drugs 2021, 81, 1683–1690. [Google Scholar] [CrossRef]
- Kitakata, H.; Endo, J.; Hashimoto, S.; Mizuno, E.; Moriyama, H.; Shirakawa, K.; Goto, S.; Katsumata, Y.; Fukuda, K.; Sano, M. Imeglimin prevents heart failure with preserved ejection fraction by recovering the impaired unfolded protein response in mice subjected to cardiometabolic stress. Biochem. Biophys. Res. Commun. 2021, 572, 185–190. [Google Scholar] [CrossRef]
- Song, H.; Tang, X.; Li, X.; Wang, Y.; Deng, A.; Wang, W.; Zhang, H.; Qin, H.; Wu, L. HLJ2 Effectively Ameliorates Colitis-Associated Cancer via Inhibition of NF-κB and Epithelial-Mesenchymal Transition. Drug Des. Dev. Ther. 2020, 14, 4291–4302. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, H.J.; Deng, A.J.; Wu, L.Q.; Zhang, Z.H.; Li, Z.H.; Wang, W.J.; Qin, H.L. Synthesis and Structure-Activity Relationships of N-Dihydrocoptisine-8-ylidene Aromatic Amines and N-Dihydrocoptisine-8-ylidene Aliphatic Amides as Antiulcerative Colitis Agents Targeting XBP1. J. Nat. Prod. 2016, 79, 775–783. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.J.; Li, Z.H.; Wu, L.Q.; Deng, A.J.; Qin, H.L. Trifluoromethylation of dihydrocoptisines and the effect on structural stability and XBP1-activating activity. J. Asian Nat. Prod. Res. 2022, 24, 388–396. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Mechanism | Ref. |
|---|---|---|
| Transcriptional Regulation | By binding to the UPRE of the Krüppel-like factor 9 (KLF9) promoter, XBP1s up-modulates the KLF9 transcription in the case of severe ER stress. | [19] |
| XBP1s induces ER expansion by activating the synthesis of phosphatidylcholine (PtdCho). | [20] | |
| XBP1s can upregulate insulin-like growth factor binding protein-3 (IGFBP3) expression. | [21] | |
| Upon ER stress, XBP1s specifically induces Forkhead Box A3 (FOXA3). FOXA3 exacerbates the excessive lipid accumulation induced by the acute ER-inducer TM. | [22] | |
| XBP1s binds to the XBP1-binding site in the Forkhead Box A2(FOXA2) promoter. | [23] | |
| XBP-1s may directly upregulate Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Thymidine Kinase (ORF21). | [24] | |
| The transcriptional activity of E1A is increased as a result of the binding of XBP1s to the E1A enhancer/promoter. | [25] | |
| XBP1s is capable of binding to and recruiting RNA polymerase II to the IL6, SNAI1, and MMP9 promoters, and the intragenic super-enhancer of glutamine-fructose-6-phosphate transaminase 2 (GFPT2). | [26] | |
| XBP1s activates MYC proto-oncogene expression. | [27] | |
| UDP-galactose-4-epimerase (GalE), a direct target of XBP1′s transcriptional function, is a new modulatory nexus that connects the UPR to the characteristic postprandial metabolic alterations in hepatocytes. | [28] | |
| The activity of the histone acetyltransferase p300 contributes to an increase in the stabilization of the spliced form of X-box binding protein 1 (XBP1s) and stimulates the transcription of XBP1′s target gene known as homocysteine inducible endoplasmic reticulum protein with ubiquitin-like domain 1 (Herpud1). | [29] | |
| Insulin Signaling | IGF-1 is responsible for inducing ER biogenesis in the bovine MEC line via the activation of the IRE1-XBP1 axis following the modulation by mTORC1. | [30] |
| Inflammation | Icariin (ICA) may reduce the expression level of TNF-α, IL-6, and IL-1β, by blocking the IRE1/XBP1s pathway. | [31] |
| The ER stress-inducing agent tunicamycin (Tm) and PA treatments significantly activate the IRE1α-XBP1s signaling pathway and increase the expression of pro-inflammatory mediators, including TNF-α, IL6, and IL1β. | [32] | |
| LncRNA H19 plays a vital function in modulating inflammation in retinal endothelial cells when subjected to high-glucose conditions via the modulation of the miR-93/XBP1s axis. | [33] | |
| The activation of the triggering receptor expressed on the myeloid cells 1 (TREM-1) pathway in macrophages causes ER stress to occur via the IRE-1α/XBP-1s pathway, thus contributing to the pro-inflammatory milieu. | [34] | |
| Oxidative stress | Complete activation of UPR, which would alleviate ER stress, is associated with fructose-induced oxidative stress. | [35] |
| Autophagy | Under PD conditions, the functional loop that governs mitophagy between XBP1s and PINK1 was disturbed. | [36] |
| ER stress/UPR is a mechanism that ultimately results in the activation of caspase-induced proteolysis and an elevation in the level of expression of genes associated with autophagy. | [37] | |
| Metabolism | XBP1s overexpression in cardiac myocytes improves the HFpEF phenotype in mice and decreases the accumulation of lipids in the myocardium. | [38] |
| XBP1s may up-modulate the expression of particular enzymes implicated in the metabolism of glucose and eventually enhance the O-linked β-N-acetylglucosamine modification (O-GlcNAcylation). | [39] | |
| Apoptosis | Inhibiting XBP1s expression might substantially improve the activities of lactate dehydrogenase and creatine kinase-MB, as well as cell apoptosis, and as a result, activated ischemia/reperfusion-mediated H9c2 cell damage is exacerbated. | [40] |
| Zonisamide (ZNS) up-modulated Bcl-2 activity, inhibited Bax and caspase-3 activity, reduced the number of TUNEL-positive cells in cardiac tissues, and protected against cardiac hypertrophy induced by type 2 diabetes by reducing the rate of apoptosis caused by ER stress (IRE-1α/XBP-1s). | [41] | |
| Grape seed proanthocyanidin extract (GSPE) relieved ER stress-elicited apoptosis via suppressing the IRE1α/XBP-1S/caspase-12 and PREK/eIF2α pathways. | [42] | |
| IRE1 made a cell-type-specific contribution to the Tg-elicited cell death. This was linked to the stimulation of c-Jun N-terminal kinase that was reliant on XBP1s, whose impact was pro-apoptotic in LNCaP cells but had no impact on HCT116 cells. | [43] | |
| STF-083010 reduced cell proliferation and induced apoptosis through XBP1/CHOP/Bim signaling pathway. | [44] | |
| Hypoxia | The stimulation of the IRE1α/XBP1s/ HIF-1α pathway was considerably inhibited when HSP47 was silenced by the use of small interfering RNA. | [45] |
| The activation of p300 by hypoxia amplifies the unfolded protein response (UPR) that is mediated by XBP-1s, helps prevent the degradation of XBP1s that is reliant on the proteasome, and increases the level of transcriptional activity carried out by XBP1s for Herpud1. | [29] | |
| The activity of IRE1 in response to hypoxia elevates the levels of the HIF1α protein in a way that is independent on XBP1s. | [46] | |
| Angiogenesis | The expression of XBP1s enhances cell proliferation, migration, and angiogenesis, which helps to reverse the damage caused by miR-33a-5p. | [47] |
| In BMECs that had been treated with OGD, XBP1s overexpression led to an increase in the expression of HIF1α, VEGF, p-extracellular signal-regulated kinase1/2, p-GSK3β, p-mTOR, p-AKT, and phosphatidylinositol-4,5-bisphosphate 3-kinase. | [48] | |
| Through the activation of XBP1s, the ER stress activator tunicamycin enhanced the production of VEGFA that was produced in human granulosa cells as a result of human chorionic gonadotropin induction. VEGFA performs a fundamental function in ovarian angiogenesis. | [49] | |
| XBP1s is responsible for the modulation of VEGF-induced angiogenesis in the heart and has a role in the development of adaptive hypertrophy. | [17] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Ding, H.; Li, Y.; Zhang, X. Molecular Mechanism Underlying Role of the XBP1s in Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 459. https://doi.org/10.3390/jcdd9120459
Liu S, Ding H, Li Y, Zhang X. Molecular Mechanism Underlying Role of the XBP1s in Cardiovascular Diseases. Journal of Cardiovascular Development and Disease. 2022; 9(12):459. https://doi.org/10.3390/jcdd9120459
Chicago/Turabian StyleLiu, Shu, Hong Ding, Yongnan Li, and Xiaowei Zhang. 2022. "Molecular Mechanism Underlying Role of the XBP1s in Cardiovascular Diseases" Journal of Cardiovascular Development and Disease 9, no. 12: 459. https://doi.org/10.3390/jcdd9120459