
Atypical Progeria Primarily Manifesting as Premature Cardiac Valvular Disease Segregates with LMNA-Gene Variants
 , , , , , and
, , , , , and 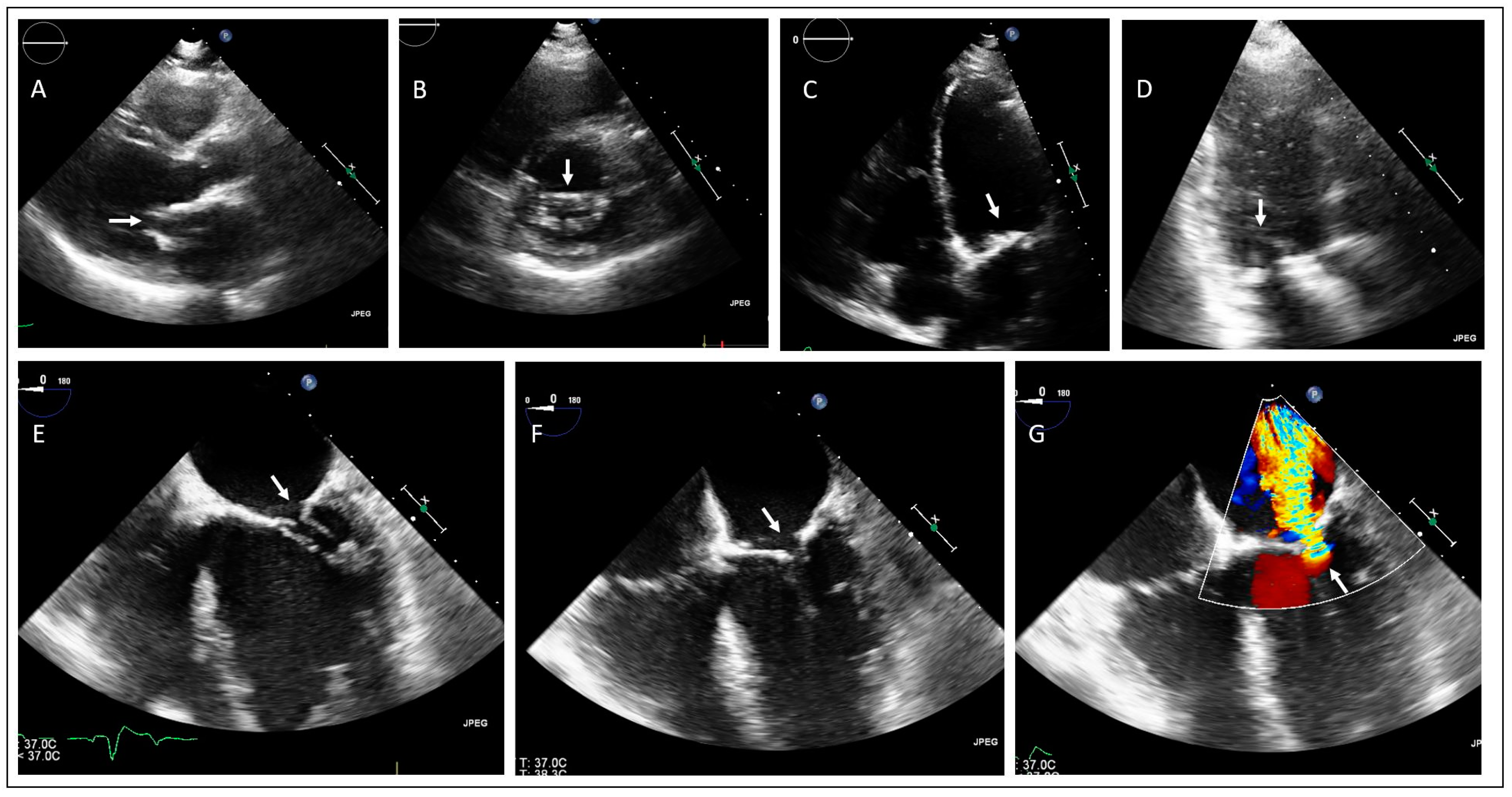{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
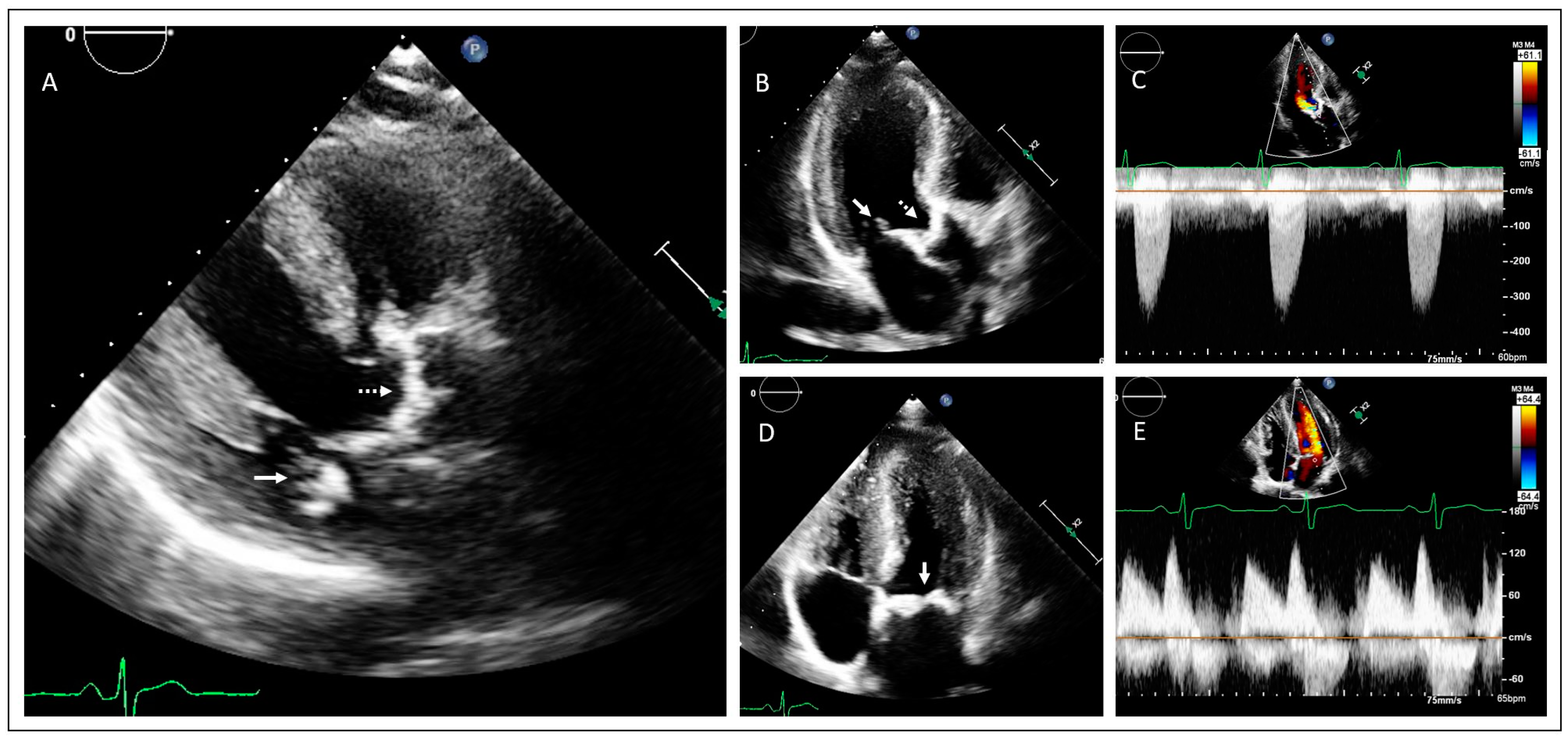
2. Case Presentations
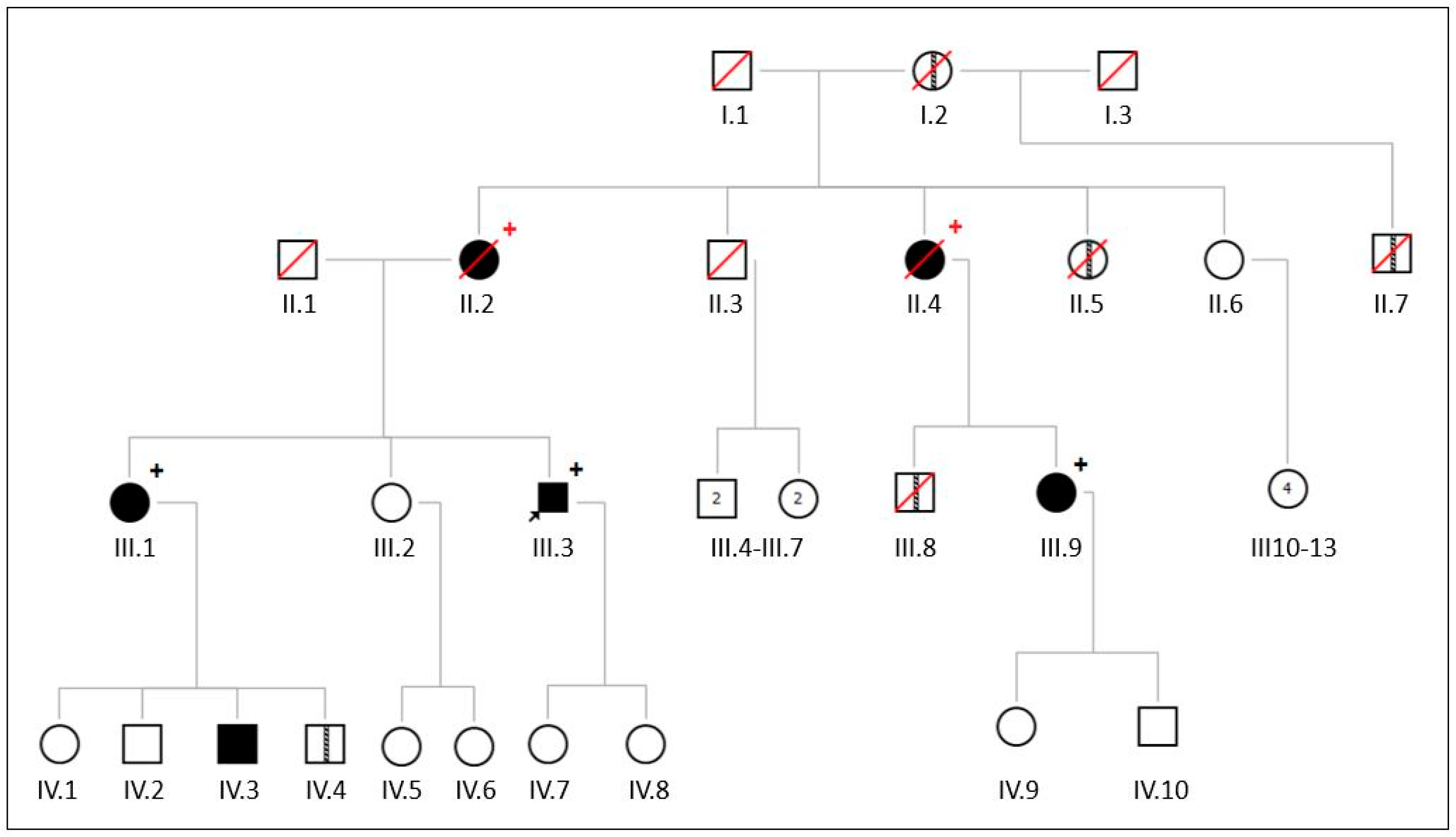
2.1. Family 1
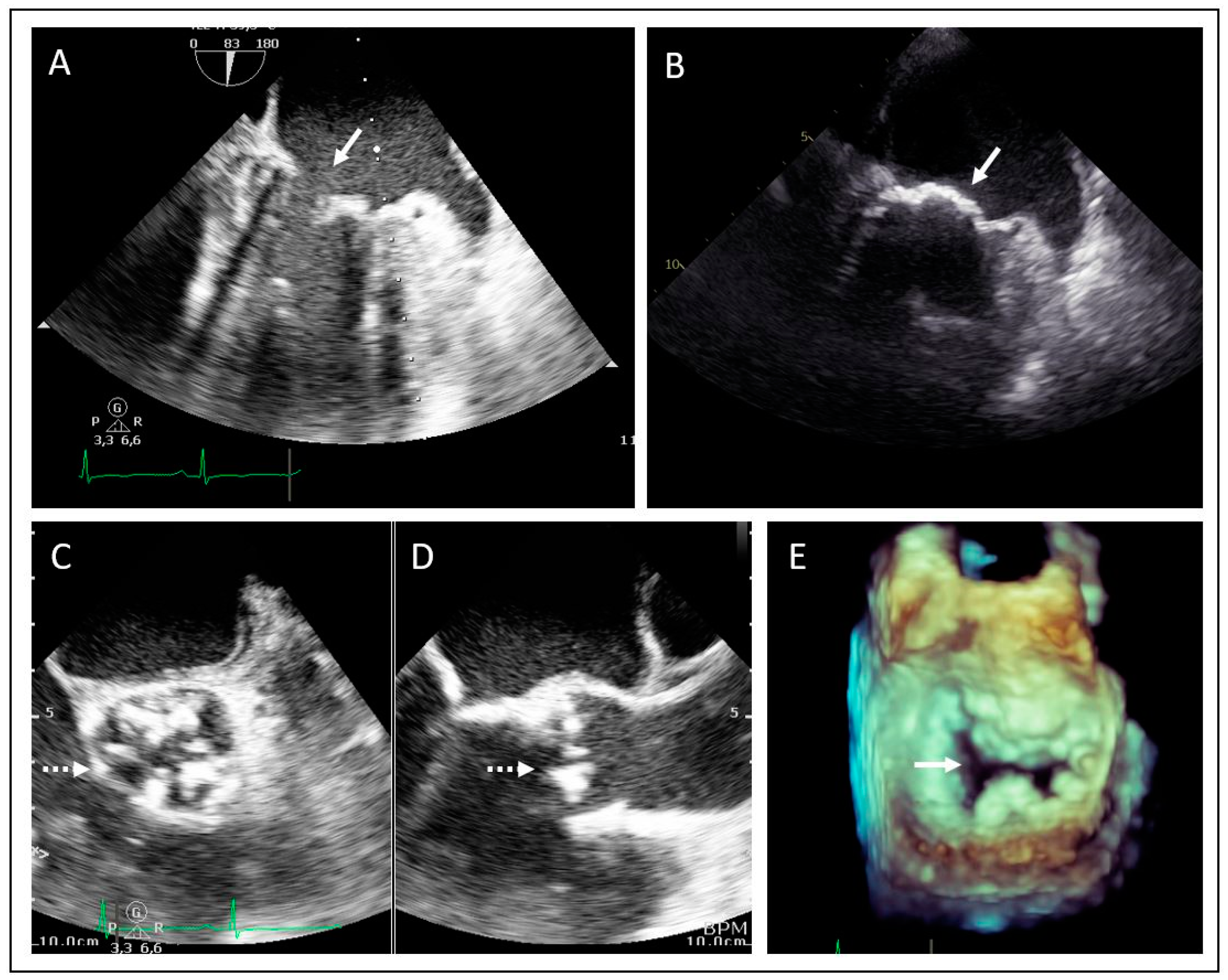
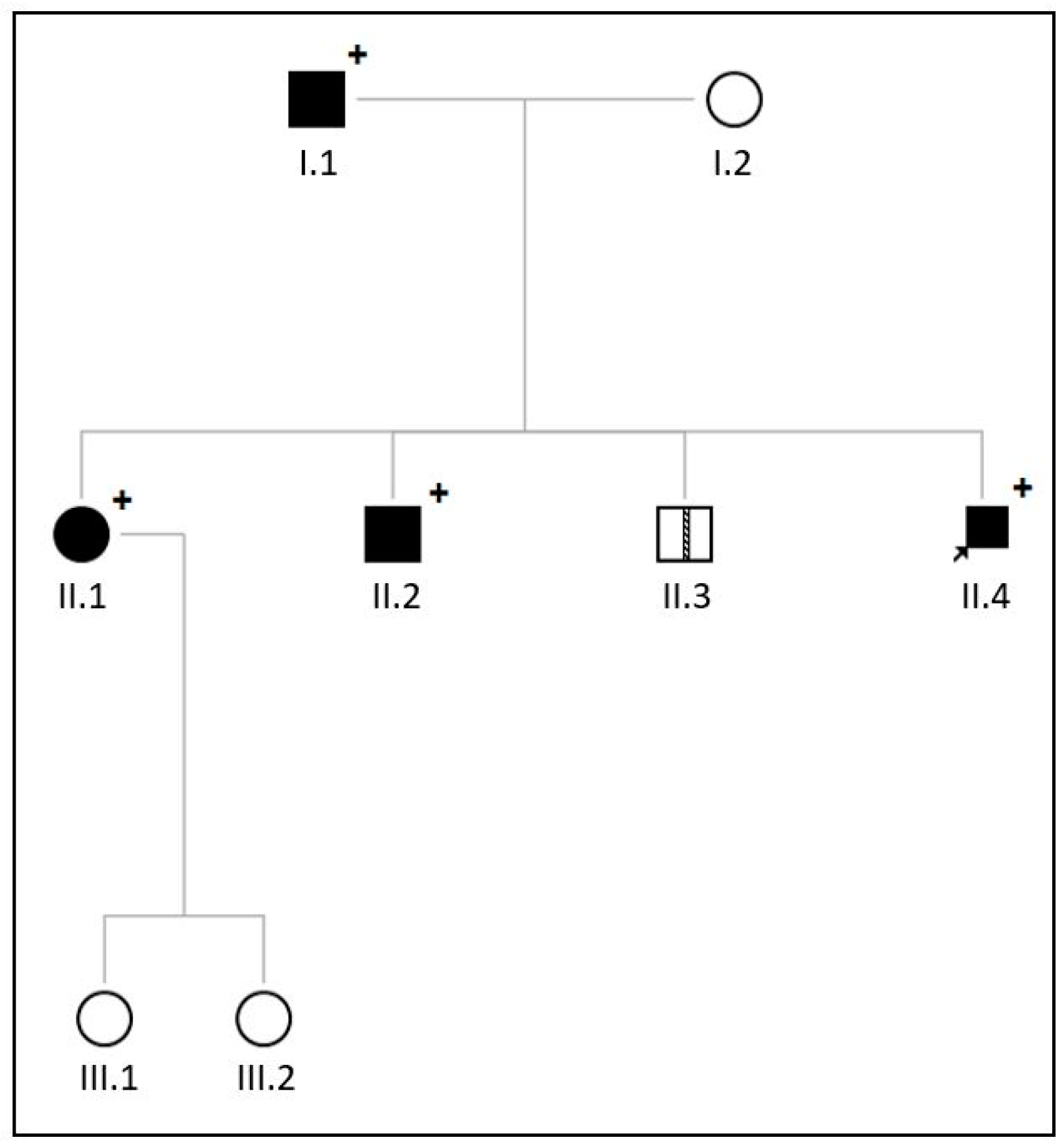
2.2. Family 2
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shin, J.Y.; Worman, H.J. Molecular Pathology of Laminopathies. Annu. Rev. Pathol. 2022, 17, 159–180. [Google Scholar] [CrossRef]
- Captur, G.; Arbustini, E.; Bonne, G.; Syrris, P.; Mills, K.; Wahbi, K.; Mohiddin, S.A.; McKenna, W.J.; Pettit, S.; Ho, C.Y.; et al. Lamin and the heart. Heart 2018, 104, 468–479. [Google Scholar] [CrossRef]
- van Berlo, J.H.; Duboc, D.; Pinto, Y.M. Often seen but rarely recognised: Cardiac complications of lamin A/C mutations. Eur. Heart J. 2004, 25, 812–814. [Google Scholar] [CrossRef]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- van Berlo, J.H.; de Voogt, W.G.; van der Kooi, A.J.; van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; de Visser, M.; et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 2005, 83, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Vidak, S.; Foisner, R. Molecular insights into the premature aging disease progeria. Histochem. Cell Biol. 2016, 145, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; Capanni, C.; Mattioli, E.; Schena, E.; Squarzoni, S.; Bacalini, M.G.; Garagnani, P.; Salvioli, S.; Franceschi, C.; Lattanzi, G. Lamin A involvement in ageing processes. Ageing Res. Rev. 2020, 62, 101073. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Yuzhalin, A.E.; Brusina, E.B.; Ponasenko, A.V.; Golovkin, A.S.; Barbarash, O.L. Genetic predisposition to calcific aortic stenosis and mitral annular calcification. Mol. Biol. Rep. 2014, 41, 5645–5663. [Google Scholar] [CrossRef] [PubMed]
- Afshar, M.; Luk, K.; Do, R.; Dufresne, L.; Owens, D.S.; Harris, T.B.; Peloso, G.M.; Kerr, K.F.; Wong, Q.; Smith, A.V.; et al. Association of Triglyceride-Related Genetic Variants With Mitral Annular Calcification. J. Am. Coll. Cardiol. 2017, 69, 2941–2948. [Google Scholar] [CrossRef] [PubMed]
- Doubaj, Y.; De Sandre-Giovannoli, A.; Vera, E.V.; Navarro, C.L.; Elalaoui, S.C.; Tajir, M.; Lévy, N.; Sefiani, A. An inherited LMNA gene mutation in atypical Progeria syndrome. Am. J. Med. Genet. A 2012, 158, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Quereda, L.; Delgadillo, V.; Juan-Mateu, J.; Verdura, E.; Rodriguez, M.J.; Baiget, M.; Pineda, M.; Gallano, P. LMNA mutation in progeroid syndrome in association with strokes. Eur. J. Med. Genet. 2011, 54, e576–e579. [Google Scholar] [CrossRef]
- Lehwald, L.M.; Renaud, D.L.; Campeau, N.G.; Babovic-Vaksanovic, D. Novel LMNA mutation in a patient with progeroid phenotype. J. Pediatr. Neurol. 2007, 5, 143–148. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Bera, M.; Ainavarapu, S.R.; Sengupta, K. Significance of 1B and 2B domains in modulating elastic properties of lamin A. Sci. Rep. 2016, 6, 27879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.A.; Scharner, J.; Felice, K.; Meriggioli, M.N.; Tarnopolsky, M.; Bower, M.; Zammit, P.S.; Mendell, J.R.; Ellis, J.A. Novel and recurrent EMD mutations in patients with Emery-Dreifuss muscular dystrophy, identify exon 2 as a mutation hot spot. J. Hum. Genet. 2011, 56, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L.; Esteves-Vieira, V.; Courrier, S.; Boyer, A.; Duong Nguyen, T.; Huong le, T.T.; Meinke, P.; Schröder, W.; Cormier-Daire, V.; Sznajer, Y.; et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur. J. Hum. Genet. 2014, 22, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Non-syndromic cardiac progeria in a patient with the rare pathogenic p.Asp300Asn variant in the LMNA gene. BMC Med. Genet. 2017, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Motegi, S.; Yokoyama, Y.; Uchiyama, A.; Ogino, S.; Takeuchi, Y.; Yamada, K.; Hattori, T.; Hashizume, H.; Ishikawa, Y.; Goto, M.; et al. First Japanese case of atypical progeroid syndrome/atypical Werner syndrome with heterozygous LMNA mutation. J. Dermatol. 2014, 41, 1047–1052. [Google Scholar] [CrossRef]
- Li, Y.; Bertozzi, A.; Mann, M.R.; Kühn, B. Interdependent changes of nuclear lamins, nuclear pore complexes, and ploidy regulate cellular regeneration and stress response in the heart. Nucleus 2023, 14, 2246310. [Google Scholar] [CrossRef]
- Perepelina, K.; Klauzen, P.; Kostareva, A.; Malashicheva, A. Tissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells. Cells 2019, 8, 266. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.W.; Van de Peppel, I.P.; Rutten, J.W.; Jukema, J.W.; Aten, E.; Jazet, I.M.; Koopmann, T.T.; Barge-Schaapveld, D.Q.C.M.; Ajmone Marsan, N. Atypical Progeria Primarily Manifesting as Premature Cardiac Valvular Disease Segregates with LMNA-Gene Variants. J. Cardiovasc. Dev. Dis. 2024, 11, 86. https://doi.org/10.3390/jcdd11030086
Wu HW, Van de Peppel IP, Rutten JW, Jukema JW, Aten E, Jazet IM, Koopmann TT, Barge-Schaapveld DQCM, Ajmone Marsan N. Atypical Progeria Primarily Manifesting as Premature Cardiac Valvular Disease Segregates with LMNA-Gene Variants. Journal of Cardiovascular Development and Disease. 2024; 11(3):86. https://doi.org/10.3390/jcdd11030086
Chicago/Turabian StyleWu, Hoi W., Ivo P. Van de Peppel, Julie W. Rutten, J. Wouter Jukema, Emmelien Aten, Ingrid M. Jazet, Tamara T. Koopmann, Daniela Q. C. M. Barge-Schaapveld, and Nina Ajmone Marsan. 2024. "Atypical Progeria Primarily Manifesting as Premature Cardiac Valvular Disease Segregates with LMNA-Gene Variants" Journal of Cardiovascular Development and Disease 11, no. 3: 86. https://doi.org/10.3390/jcdd11030086