

Unexplained Left Ventricular Hypertrophy Diagnosed as a Cardiac Variant of Late-Onset Fabry Disease: A Case Report
 , ,
, ,
Abstract
:1. Introduction
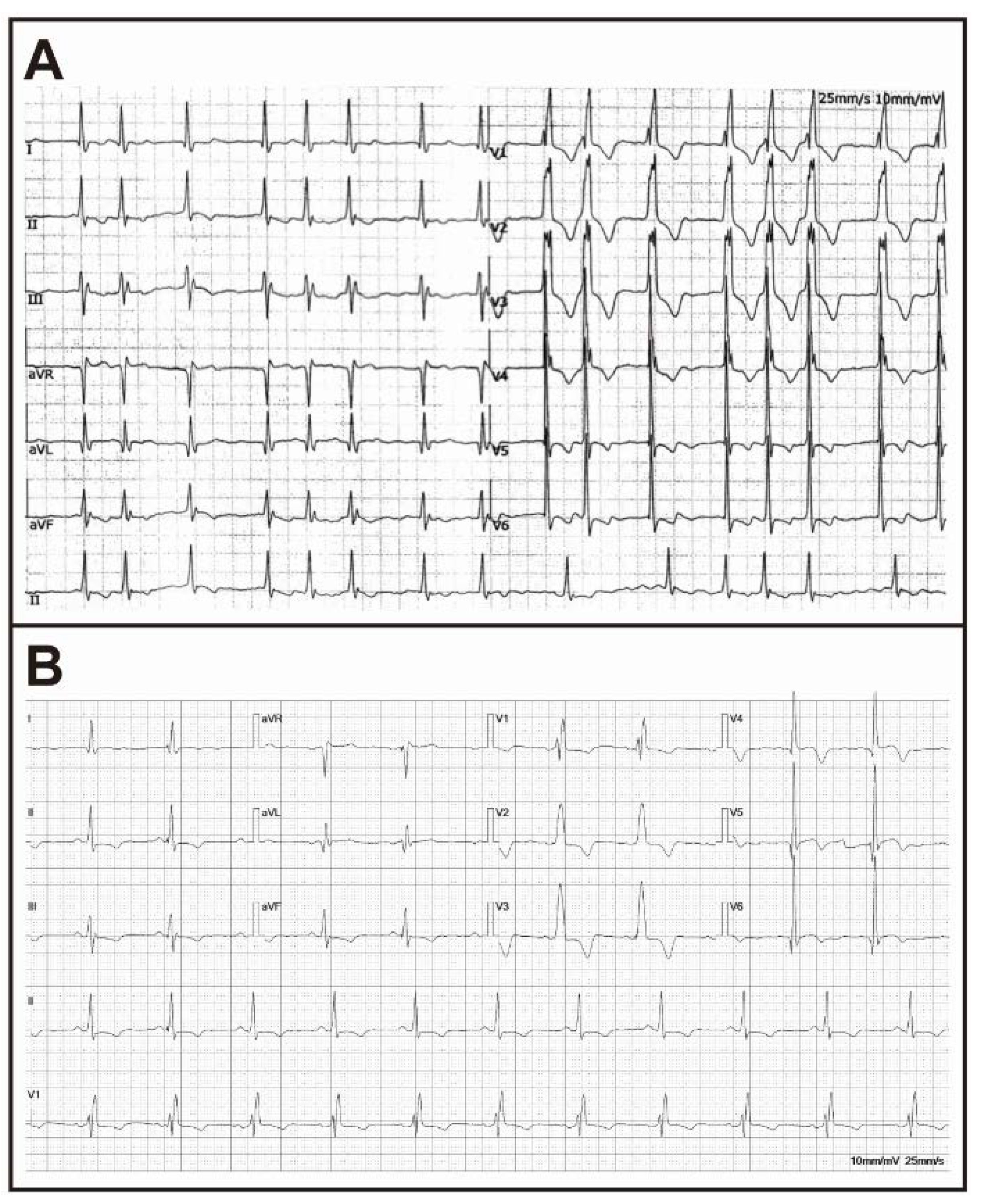
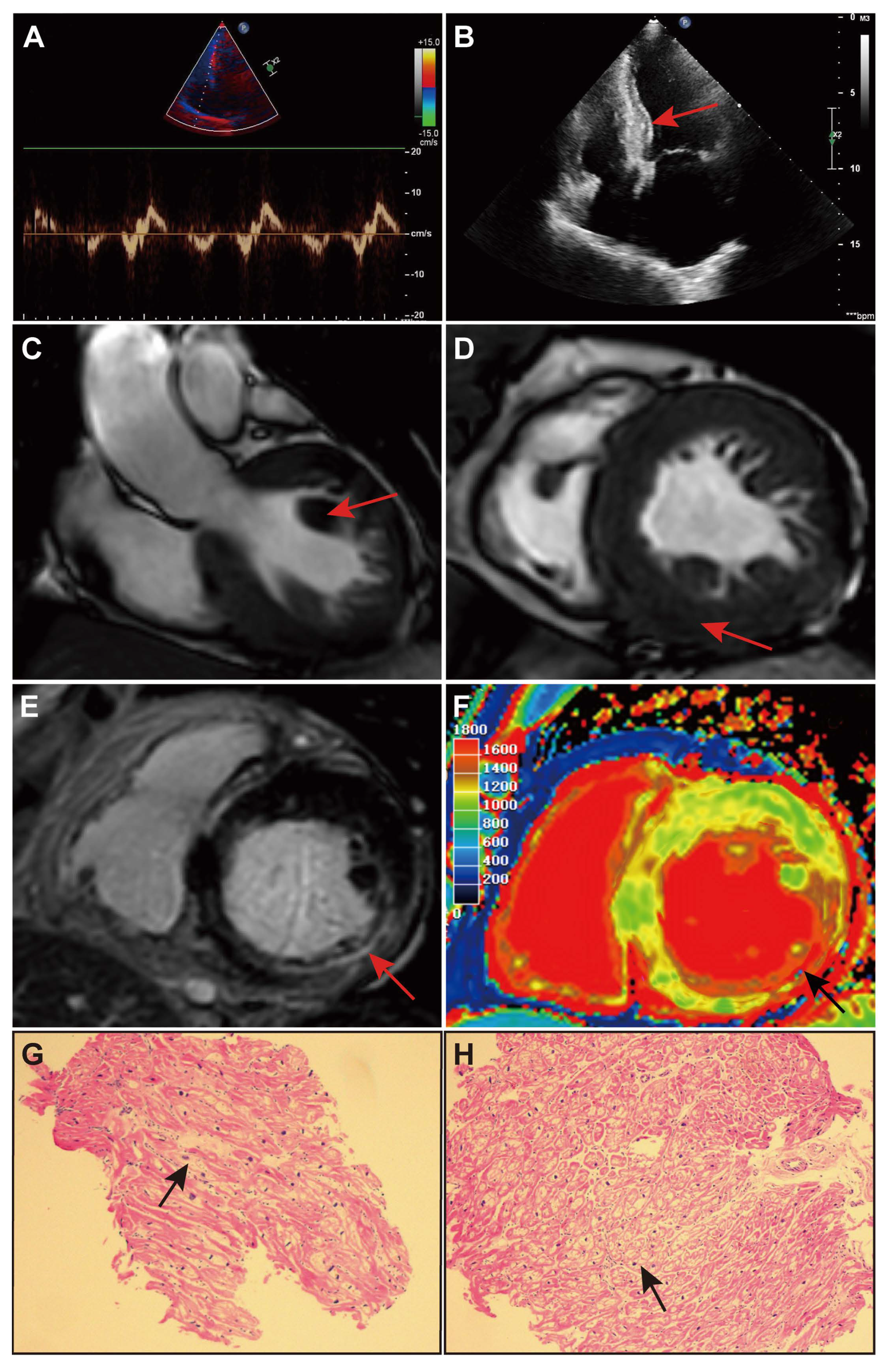
2. Case Presentation
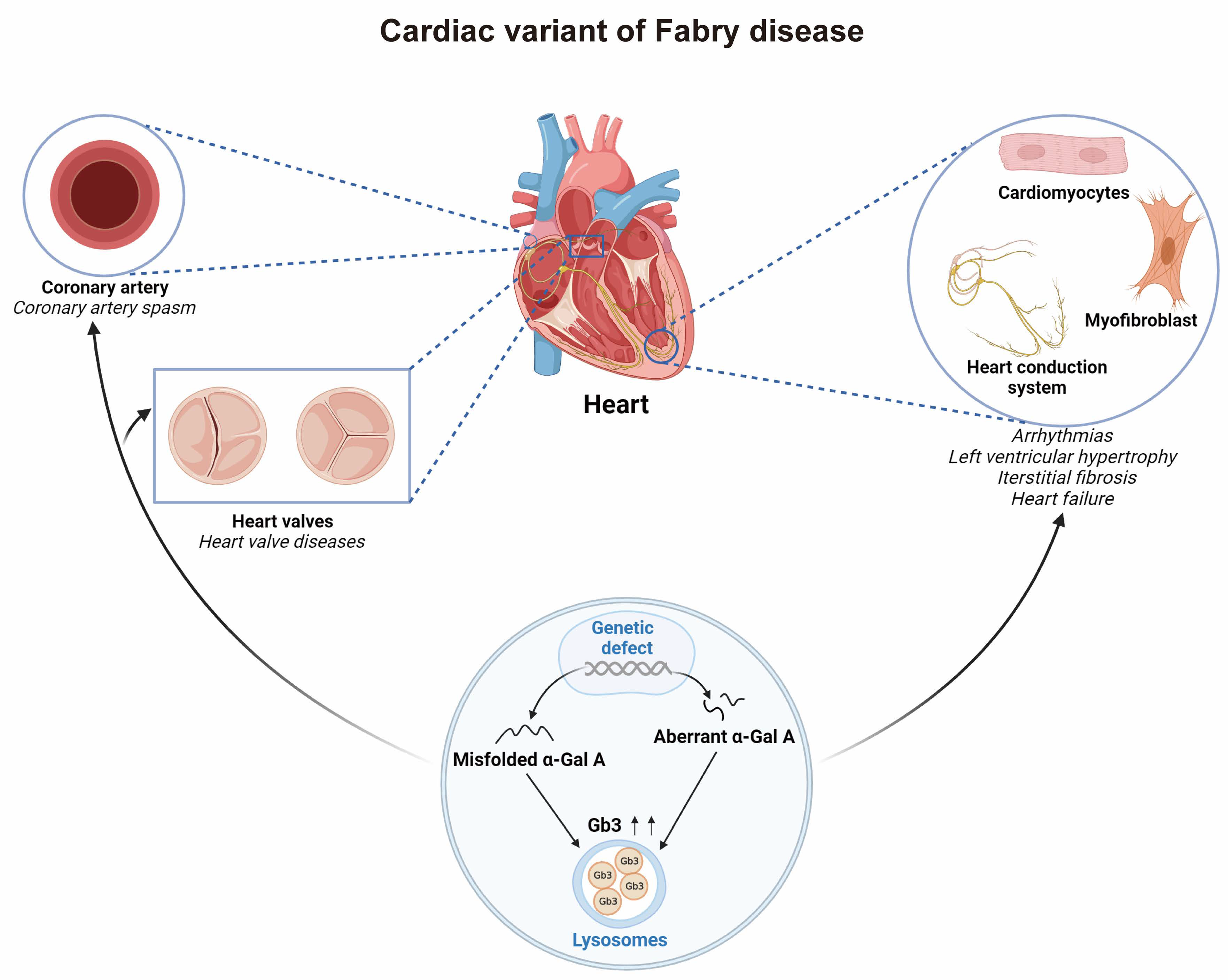
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry Disease Revisited: Management and Treatment Recommendations for Adult Patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, M.N.; Cane, P.; Florio, R.; Kavantzas, N.; Close, L.; Shah, J.; Lee, P.; Elliott, P. A Detailed Pathologic Examination of Heart Tissue from Three Older Patients with Anderson-Fabry Disease on Enzyme Replacement Therapy. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2010, 19, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Brand, E. Precision Medicine in Fabry Disease. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2021, 36, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Niemann, M.; Störk, S.; Breunig, F.; Beer, M.; Sommer, C.; Herrmann, S.; Ertl, G.; Wanner, C. Long-Term Outcome of Enzyme-Replacement Therapy in Advanced Fabry Disease: Evidence for Disease Progression towards Serious Complications. J. Intern. Med. 2013, 274, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Bacharova, L. ECG in Left Ventricular Hypertrophy: A Change in Paradigm from Assessing Left Ventricular Mass to Its Electrophysiological Properties. J. Electrocardiol. 2022, 73, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Sheikh, N.; Biagini, E.; Papadakis, M.; Maurizi, N.; Sinagra, G.; Pelliccia, A.; Rapezzi, C.; Sharma, S.; Olivotto, I. The Electrocardiogram in the Diagnosis and Management of Patients with Hypertrophic Cardiomyopathy. Heart Rhythm. 2020, 17, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Militaru, S.; Ginghină, C.; Popescu, B.A.; Săftoiu, A.; Linhart, A.; Jurcuţ, R. Multimodality Imaging in Fabry Cardiomyopathy: From Early Diagnosis to Therapeutic Targets. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Brand, E. Fabry Disease: The Current Treatment Landscape. Drugs 2021, 81, 635–645. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Year | Age | Symptoms | Evaluation | Main Diagnosis | Management |
|---|---|---|---|---|---|
| 2019 | 64 | Chest pain | ECG: Atypical right bundle branch block CAG: Negative TTE: Reduced rate of contraction and thickening of the lower left ventricular wall LVEF = 62% | Acute myocardial infarction and coronary artery spasm | AMI drugs, outpatient follow-up |
| 2021 | 66 | Chest pain and palpitation | ECG: Atrial fibrillation and atypical right bundle branch block TTE: Left ventricular hypertrophy and LV diastolic dysfunction LVEF = 59% | Acute myocardial infarction, coronary artery spasm, and atrial fibrillation | AF drugs, NDHP-CCB, and outpatient follow-up |
| 2023 | 68 | Palpitation and edema of both lower limbs | ECG: Atypical right bundle branch block, atrial fibrillation, and T-wave inversion TTE: LVH, LV diastolic dysfunction, enlarged left atrial and right atrial internal diameters LVEF = 63% CMR: Hypertrophic cardiomyopathy with diffuse lipid infiltration of the myocardium | Fabry disease, atrial fibrillation, and diastolic heart failure | ERT, AF drugs, HF drugs, and outpatient follow-up |
| Examination | Results | Reference Value |
|---|---|---|
| α-Gal A activity | <1 nmol/1 h/mg | N > 24.70 (nmol/1 h/mg) |
| Endomyocardial biopsy | Most cardiomyocytes are vacuolated | Compatible with FD |
| Genetic sequencing | p.Arg301Gln (c.902G > C) heterozygous variant encoding exon 6 of the GLA gene (NM_000169) | Compatible with FD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Niu, X.; Bai, L.; Zhang, Y.; Wang, T.; Wa, Y.; Wei, J.; Dong, K.; Zhang, X.; Bai, M. Unexplained Left Ventricular Hypertrophy Diagnosed as a Cardiac Variant of Late-Onset Fabry Disease: A Case Report. J. Cardiovasc. Dev. Dis. 2023, 10, 389. https://doi.org/10.3390/jcdd10090389
Zhao M, Niu X, Bai L, Zhang Y, Wang T, Wa Y, Wei J, Dong K, Zhang X, Bai M. Unexplained Left Ventricular Hypertrophy Diagnosed as a Cardiac Variant of Late-Onset Fabry Disease: A Case Report. Journal of Cardiovascular Development and Disease. 2023; 10(9):389. https://doi.org/10.3390/jcdd10090389
Chicago/Turabian StyleZhao, Maomao, Xiaowei Niu, Lu Bai, Yinchang Zhang, Ting Wang, Yongling Wa, Junchu Wei, Kang Dong, Xin Zhang, and Ming Bai. 2023. "Unexplained Left Ventricular Hypertrophy Diagnosed as a Cardiac Variant of Late-Onset Fabry Disease: A Case Report" Journal of Cardiovascular Development and Disease 10, no. 9: 389. https://doi.org/10.3390/jcdd10090389