

The Intriguing Role of Hypoxia-Inducible Factor in Myocardial Ischemia and Reperfusion: A Comprehensive Review

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hypoxia-Inducible Factors (HIFs)—An Overview
2.1. Structure of HIFs
2.2. Regulation of HIFs under Hypoxic Conditions
2.3. Role of HIFs in Various Physiological Processes
3. Myocardial Ischemia and Reperfusion Injury: Pathophysiology
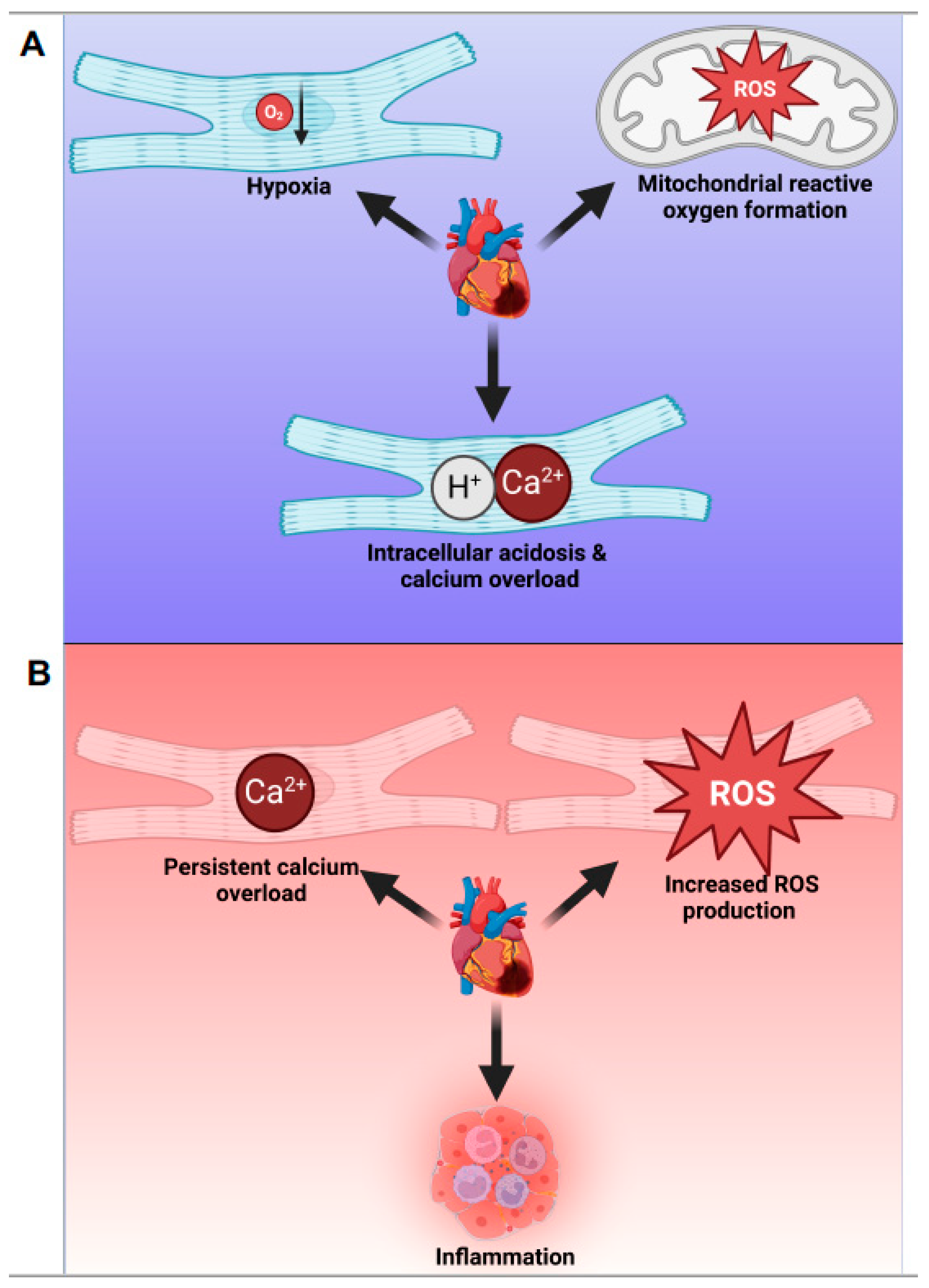
3.1. Cellular and Molecular Events during Myocardial Ischemia
3.2. Cellular and Molecular Events during Reperfusion Injury
4. Role of HIFs in Myocardial Ischemia and Reperfusion Injury
4.1. Activation of HIF-Isoforms during Ischemia and Reperfusion
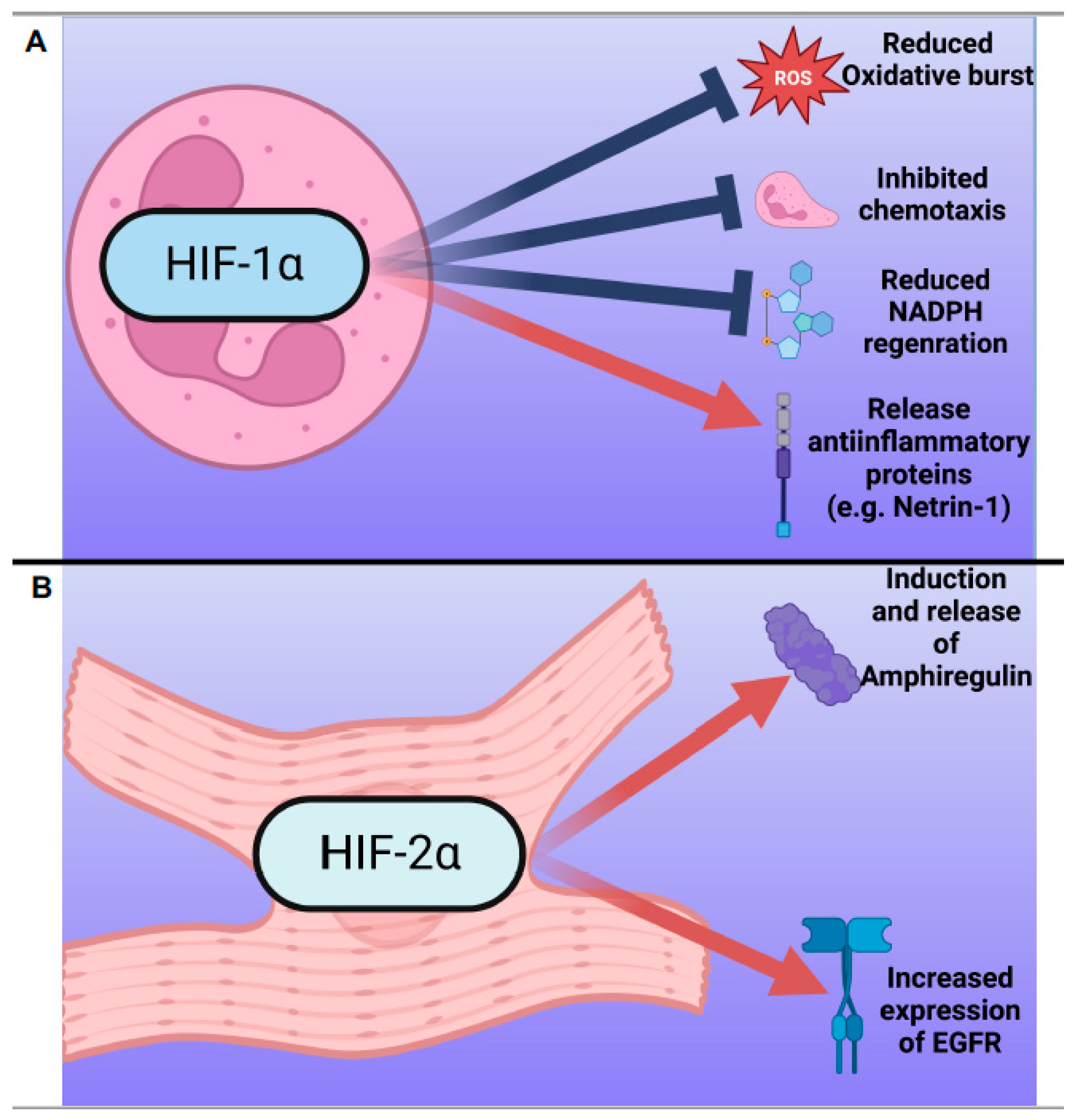
4.1.1. HIF-1α
4.1.2. HIF-2α
4.2. Differential Sensitivity of HIF-1α and HIF-2α to Oxygen Partial Pressure in Myocardial Ischemia and Reperfusion
5. Pharmacological Modulation of HIFs in Myocardial Ischemia and Reperfusion Injury
5.1. HIF Stabilizers in Clinical Trials
5.2. Challenges and Limitations of Pharmacological HIF Modulation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turer, A.T.; Hill, J.A. Pathogenesis of Myocardial Ischemia-Reperfusion Injury and Rationale for Therapy. Am. J. Cardiol. 2010, 106, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Prize, N. The Nobel Prize in Physiology or Medicine 2019—Summary. Resonance 2019, 24, 1375–1380. [Google Scholar]
- Smith, T.G.; Robbins, P.; Ratcliffe, P. The human side of hypoxia-inducible factor. Br. J. Haematol. 2008, 141, 325–334. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1 alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef]
- Makino, Y.; Uenishi, R.; Okamoto, K.; Isoe, T.; Hosono, O.; Tanaka, H.; Kanopka, A.; Poellinger, L.; Haneda, M.; Morimoto, C. Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1): A negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. J. Biol. Chem. 2007, 282, 14073–14082. [Google Scholar] [CrossRef]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of Oxygen Signaling at the Consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Schödel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and Mammalian Homologs Define a Family of Dioxygenases that Regulate HIF by Prolyl Hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes. Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Egginton, S. Invited review: Activity-induced angiogenesis. Pflug. Arch. 2009, 457, 963–977. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, H.E.; Chiappe, G.E.; Ennis, I.L.; Morgan, P.G.; Alvarez, B.V.; Casey, J.R.; Camilión de Hurtado, M.C. Influence of Na+-independent Cl-HCO3- exchange on the slow force response to myocardial stretch. Circ. Res. 2003, 93, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Orchard, C.H.; Kentish, J.C.; Saegusa, N.; Garg, V.; Spitzer, K.W.; Parker, M.D.; Boron, W.F.; Cingolani, H.E.; Pérez, N.G.; Cingolani, O.H.; et al. Effects of changes of pH on the contractile function of cardiac muscle. Am. J. Physiol. Physiol. 1990, 258, C967–C981. [Google Scholar] [CrossRef]
- Carmeliet, P.; Baes, M. Metabolism and Therapeutic Angiogenesis. N. Engl. J. Med. 2008, 358, 2511–2512. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell. Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Jugdutt, B.I. Ventricular remodeling after infarction and the extracellular collagen matrix: When is enough enough? Circulation 2003, 108, 1395–1403. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Zweier, J.L.; Talukder, M.A.H. The role of oxidants and free radicals in reperfusion injury. Cardiovasc. Res. 2006, 70, 181–190. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell. Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Eisner, D.; Nichols, C.G.; O’Neill, S.C.; Smith, G.L.; Valdeolmillos, M. The effects of metabolic inhibition on intracellular calcium and pH in isolated rat ventricular cells. J. Physiol. 1989, 411, 393–418. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium Fluxes Involved in Control of Cardiac Myocyte Contraction. Circ. Res. 2000, 87, 275–281. [Google Scholar] [CrossRef]
- Allen, D.G.; Orchard, C.H. Myocardial contractile function during ischemia and hypoxia. Circ. Res. 1987, 60, 153–168. [Google Scholar] [CrossRef]
- Jennings, R.B.; Sommers, H.M.; Smyth, G.A.; Flack, H.A.; Linn, H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch. Pathol. 1960, 70, 68–78. [Google Scholar]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reper-fusion--a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef]
- Di Lisa, F.; Bernardi, P. Mitochondria and ischemia–reperfusion injury of the heart: Fixing a hole. Cardiovasc. Res. 2006, 70, 191–199. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Jordan, J.E.; Zhao, Z.-Q.; Vinten-Johansen, J. The role of neutrophils in myocardial ischemia–reperfusion injury. Cardiovasc. Res. 1999, 43, 860–878. [Google Scholar] [CrossRef] [PubMed]
- Vakeva, A.P.; Agah, A.; Rollins, S.A.; Matis, L.A.; Li, L.; Stahl, G.L. Myocardial Infarction and Apoptosis After Myocardial Ischemia and Reperfusion: Role of the Terminal Complement Components and Inhibition by Anti-C5 Therapy. Circulation 1998, 97, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Entman, M.L.; Youker, K.; Shoji, T.; Kukielka, G.; Shappell, S.B.; Taylor, A.; Smith, C.W. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. J. Clin. Investig. 1992, 90, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Romson, J.L.; Hook, B.G.; Kunkel, S.L.; Abrams, G.D.; A Schork, M.; Lucchesi, B.R. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 1983, 67, 1016–1023. [Google Scholar] [CrossRef]
- Kukielka, G.L.; Smith, C.W.; LaRosa, G.J.; Manning, A.M.; Mendoza, L.H.; Daly, T.J.; Hughes, B.J.; Youker, K.; Hawkins, H.K.; Michael, L.H. Interleukin-8 gene induction in the myocardium after ischemia and reperfusion in vivo. J. Clin. Investig. 1995, 95, 89–103. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Heck-Swain, K.L.; Li, J.; Ruan, W.; Yuan, X.; Wang, Y.; Koeppen, M.; Eltzschig, H.K. Myeloid hypoxia-inducible factor HIF1A provides cardio-protection during ischemia and reperfusion via induction of netrin-1. Front. Cardiovasc. Med. 2022, 9, 2553. [Google Scholar] [CrossRef]
- Elks, P.; Van Eeden, F.J.; Dixon, G.; Wang, X.; Reyes-Aldasoro, C.C.; Ingham, P.W.; Whyte, M.K.B.; Walmsley, S.; Renshaw, S.A. Activation of hypoxia-inducible factor-1α (Hif-1α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood 2011, 118, 712–722. [Google Scholar] [CrossRef]
- Lin, N.; Simon, M.C. Hypoxia-inducible factors: Key regulators of myeloid cells during inflammation. J. Clin. Investig. 2016, 126, 3661–3671. [Google Scholar] [CrossRef]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1{alpha} expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest. 2005, 115, 1806–1815. [Google Scholar] [CrossRef]
- Jun, H.S.; Weinstein, D.A.; Lee, Y.M.; Mansfield, B.C.; Chou, J.Y. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood 2014, 123, 2843–2853. [Google Scholar] [CrossRef]
- Walmsley, S.R.; Whyte, M.K. Neutrophil energetics and oxygen sensing. Blood 2014, 123, 2753–2754. [Google Scholar] [CrossRef]
- Pérez-Ladaga, A.; Muñoz, M.; Mastora, C.; Sola, A. HIF-1α Provokes Delayed Neutrophil Apoptosis by Decreasing 24P3 Expression and Intracellular Iron Content. Eur. J. Inflamm. 2014, 12, 53–65. [Google Scholar] [CrossRef]
- Rosenberger, P.; Schwab, J.M.; Mirakaj, V.; Masekowsky, E.; Mager, A.; Morote-Garcia, J.C.; Unertl, K.; Eltzschig, H.K. Hypoxia-inducible factor–dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 2009, 10, 195–202. [Google Scholar] [CrossRef]
- Li, J.; Conrad, C.; Mills, T.W.; Berg, N.K.; Kim, B.; Ruan, W.; Lee, J.W.; Zhang, X.; Yuan, X.; Eltzschig, H.K. PMN-derived netrin-1 attenuates cardiac ischemia-reperfusion injury via myeloid ADORA2B signaling. J. Exp. Med. 2021, 218, e20210008. [Google Scholar] [CrossRef]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef]
- Xiao, J.; Lv, Y.; Lin, B.; Tipoe, G.L.; Youdim, M.B.; Xing, F.; Liu, Y. A Novel Antioxidant Multitarget Iron Chelator M30 Protects Hepatocytes Against Ethanol-Induced Injury. Oxidative Med. Cell. Longev. 2015, 2015, 607271. [Google Scholar] [CrossRef]
- Sherman, M.A.; Suresh, M.V.; Dolgachev, V.A.; McCandless, L.K.; Xue, X.; Ziru, L.; Machado-Aranda, D.; Shah, Y.M.; Raghavendran, K. Molecular Characterization of Hypoxic Alveolar Epithelial Cells After Lung Contusion Indicates an Important Role for HIF-1α. Ann. Surg. 2018, 267, 382. [Google Scholar] [CrossRef]
- Ortiz, V.D.; Teixeira, R.B.; Türck, P.; Corssac, G.B.; Belló-Klein, A.; de Castro, A.L.; Araujo, A.S.D.R. Influence of Carvedilol and Thyroid Hormones on Inflammatory Proteins and Cardioprotective Factor HIF-1α in the Infarcted Heart. Can. J. Physiol. Pharmacol. 2023, 10, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.J.; Liu, T.; Yang, H.H.; Duan, J.X.; Yang, J.T.; Guan, X.X.; Xiong, J.B.; Zhang, Y.F.; Zhang, C.Y.; Zhou, Y.; et al. TREM-1 Governs NLRP3 Inflammasome Activation of Macrophages by Firing Up Glycolysis in Acute Lung Injury. Int. J. Biol. Sci. 2023, 19, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Sant’Ana, P.G.; Tomasi, L.C.D.; Murata, G.M.; Vileigas, D.F.; Mota, G.A.F.; Souza, S.L.B.D.; Silva, V.L.; Campos, L.P.D.; Okoshi, K.; Padovani, C.R.; et al. Hypoxia-Inducible Factor 1-Alpha and Glucose Metabolism During Cardiac Remodeling Progression From Hypertrophy to Heart Failure. Int. J. Mol. Sci. 2023, 24, 6201. [Google Scholar] [CrossRef] [PubMed]
- Lan, G.; Batu, L.; Guang, L. Hypoxia Induces Autophagy in Cardiomyocytes via a Hypoxia-Inducible Factor 1-Dependent Mechanism. Exp. Ther. Med. 2016, 11, 2233–2239. [Google Scholar]
- Belaidi, E.; Beguin, P.C.; Levy, P.; Ribuot, C.; Godin-Ribuot, D. Prevention of HIF-1 Activation and iNOS Gene Targeting by Low-Dose Cadmium Results in Loss of Myocardial Hypoxic Preconditioning in the Rat. AJP Heart Circ. Physiol. 2008, 294, H901–H908. [Google Scholar] [CrossRef]
- Li, X.; Zhao, H.; Wu, Y.; Zhang, S.; Zhao, X.; Zhang, Y.; Wang, J.; Wang, J.; Liu, H. Up-Regulation of Hypoxia-Inducible Factor-1α Enhanced the Cardioprotective Effects of Ischemic Postconditioning in Hyperlipidemic Rats. Acta Biochim. Biophys. Sin. 2014, 46, 112–118. [Google Scholar] [CrossRef]
- Demet, T.; Ali, D.; Lei, X. Hypoxia Inducible Factor 1 (HIF-1) and Cardioprotection. Acta Pharmacol. Sin. 2010, 31, 1085–1094. [Google Scholar]
- Ong, S.-G.; Lee, W.H.; Theodorou, L.; Kodo, K.; Lim, S.Y.; Shukla, D.H.; Briston, T.; Kiriakidis, S.; Ashcroft, M.; Davidson, S.M.; et al. HIF-1 reduces ischaemia–reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc. Res. 2014, 104, 24–36. [Google Scholar] [CrossRef]
- Kasivisvanathan, V.; Shalhoub, J.; Lim, C.S.; Shepherd, A.C.; Thapar, A.; Davies, A.H. Hypoxia-Inducible Factor-1 in Arterial Disease: A Putative Therapeutic Target. Curr. Vasc. Pharmacol. 2011, 9, 333–349. [Google Scholar] [CrossRef]
- Zheng, Z.L.; Hwang, Y.H.; Kim, S.K.; Kim, S.; Son, M.J.; Ro, H.; Sung, S.A.; Lee, H.H.; Chung, W.K.; Joo, K.W.; et al. Genetic Polymorphisms of Hypoxia-Inducible Factor-1 Alpha and Cardiovascular Disease in Hemodialysis Patients. Nephron Clin. Pract. 2009, 113, c104–c111. [Google Scholar] [CrossRef]
- Minamishima, Y.A.; Moslehi, J.; Bardeesy, N.; Cullen, D.; Bronson, R.T.; Kaelin, W.G., Jr. Somatic Inactivation of the PHD2 Prolyl Hydroxylase Causes Polycythemia and Congestive Heart Failure. Blood 2008, 111, 3236–3244. [Google Scholar] [CrossRef]
- Kido, M.; Du, L.; Sullivan, C.C.; Li, X.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Hypoxia-Inducible Factor 1-Alpha Reduces Infarction and Attenuates Progression of Cardiac Dysfunction After Myocardial Infarction in the Mouse. J. Am. Coll. Cardiol. 2005, 46, 2116–2124. [Google Scholar] [CrossRef]
- Wu, J.; Yang, L.; Xie, P.; Yu, J.; Yu, T.; Wang, H.; Maimaitili, Y.; Wang, J.; Ma, H.; Yang, Y.; et al. Cobalt Chloride Upregulates Impaired HIF-1α Expression to Restore Sevoflurane Post-Conditioning-Dependent Myocardial Protection in Diabetic Rats. Front. Physiol. 2017, 8, 395. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, J.; Wu, J.; Gu, T.W.; Ti, T.L.; Chen, S. Activating Hypoxia-Inducible Factor-1α Reduces Myocardial Ischemia-Reperfusion Injury in Mice Through Hexokinase II. J. Biomater. Tissue Eng. 2022, 12, 1626–1635. [Google Scholar] [CrossRef]
- Nordquist, L.; Friederich-Persson, M.; Fasching, A.; Liss, P.; Shoji, K.; Nangaku, M.; Hansell, P.; Palm, F. Activation of Hypoxia-Inducible Factors Prevents Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 328–338. [Google Scholar] [CrossRef]
- Shohet, R.V.; Garcia, J.A. Keeping the engine primed: HIF factors as key regulators of cardiac metabolism and angiogenesis during ischemia. J. Mol. Med. 2007, 85, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Liu, Y.; Zhao, J.; Cai, L.; Li, X.; Feng, W. Activation of HIF-1 by Metallothionein Contributes to Cardiac Protection in the Diabetic Heart. AJP Heart Circ. Physiol. 2012, 302, H2528–H2535. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ichise, N.; Kobayashi, T.; Fusagawa, H.; Yamazaki, H.; Kudo, T.; Tohse, N. Enhanced Glucose Metabolism Through Activation of HIF-1α Covers the Energy Demand in a Rat Embryonic Heart Primordium After Heartbeat Initiation. Sci. Rep. 2022, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Bohuslavova, R.; Kolar, F.; Sedmera, D.; Skvorova, L.; Papousek, F.; Neckar, J.; Pavlinkova, G. Partial Deficiency of HIF-1α Stimulates Pathological Cardiac Changes in Streptozotocin-Induced Diabetic Mice. BMC Endocr. Disord. 2014, 14, 11. [Google Scholar] [CrossRef]
- Jiang, X.; Tian, W.; Tu, A.B.; Pasupneti, S.; Shuffle, E.; Dahms, P.; Zhang-Benoit, Y.; Cai, H.; Dinh, T.T.; Li, R.-J.; et al. Endothelial Hypoxia-Inducible Factor-2α Is Required for the Maintenance of Airway Microvasculature. Circulation 2019, 139, 502–517. [Google Scholar] [CrossRef]
- Koeppen, M.; Lee, J.W.; Seo, S.W.; Brodsky, K.S.; Kreth, S.; Yang, I.V.; Buttrick, P.M.; Eckle, T.; Eltzschig, H.K. Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ische-mia-reperfusion injury. Nat. Commun. 2018, 9, 816. [Google Scholar] [CrossRef]
- Mastrocola, R.; Collino, M.; Penna, C.; Nigro, D.; Chiazza, F.; Fracasso, V.; Tullio, F.; Alloatti, G.; Pagliaro, P.; Aragno, M. Maladaptive Modulations of NLRP3 Inflammasome and Cardioprotective Pathways Are Involved in Diet-Induced Exacerbation of Myocardial Ischemia/Reperfusion Injury in Mice. Oxidative Med. Cell. Longev. 2015, 2016, 3480637. [Google Scholar] [CrossRef]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443. [Google Scholar] [CrossRef]
- Zhang, S.; Han, C.-H.; Chen, X.-S.; Zhang, M.; Xu, L.-M.; Zhang, J.-J.; Xia, Q. Transient Ureteral Obstruction Prevents against Kidney Ischemia/Reperfusion Injury via Hypoxia-Inducible Factor (HIF)-2α Activation. PLoS ONE 2012, 7, e29876. [Google Scholar] [CrossRef]
- Cowburn, A.S.; Takeda, N.; Boutin, A.T.; Kim, J.W.; Sterling, J.C.; Nakasaki, M.; Southwood, M.; Goldrath, A.W.; Jamora, C.; Nizet, V.; et al. HIF isoforms in the skin differentially regulate systemic arterial pressure. Proc. Natl. Acad. Sci. USA 2013, 110, 17570–17575. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Lee, J.W.; Koeppen, M.; Seo, S.W.; Bowser, J.L.; Yuan, X.; Li, J.; Sibilia, M.; Ambardekar, A.V.; Zhang, X.; Eckle, T.; et al. Transcription-independent Induction of ERBB1 through Hypoxia-inducible Factor 2A Provides Cardioprotec-tion during Ischemia and Reperfusion. Anesthesiology 2020, 132, 763–780. [Google Scholar] [CrossRef]
- Chen, Y.-R.; Dai, A.-G.; Hu, R.-C.; Jiang, Y.-L. Differential and Reciprocal Regulation between Hypoxia-inducible Factor-α Subunits and their Prolyl Hydroxylases in Pulmonary Arteries of Rat with Hypoxia-induced Hypertension. Acta Biochim. Biophys. Sin. 2006, 38, 423–434. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Kochan, K.; Piotrowski, A.; Kamysz, W.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. The hypoxia-inducible miR-429 regulates hypoxia-inducible factor-1α expression in human endothelial cells through a negative feedback loop. FASEB J. 2015, 29, 1467–1479. [Google Scholar] [CrossRef]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: Implication of natural antisense HIF-1alpha. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Hamidian, A.; von Stedingk, K.; Thorén, M.M.; Mohlin, S.; Påhlman, S. Differential regulation of HIF-1α and HIF-2α in neuroblastoma: Estrogen-related receptor alpha (ERRα) regulates HIF2A transcription and correlates to poor outcome. Biochem. Biophys. Res. Commun. 2015, 461, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Downes, N.L.; Laham-Karam, N.; Kaikkonen, M.U.; Ylä-Herttuala, S. Differential but Complementary HIF1α and HIF2α Transcriptional Regulation. Mol. Ther. 2018, 26, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shi, S.; Shi, Q.; Zhang, H.; Hu, R.; Wang, M. Similarity in the functions of HIF-1α and HIF-2α proteins in cervical cancer cells. Oncol. Lett. 2017, 14, 5643–5651. [Google Scholar] [CrossRef] [PubMed]
- Vukovic, M.; Guitart, A.V.; Sepulveda, C.; Villacreces, A.; O’Duibhir, E.; Panagopoulou, T.I.; Ivens, A.; Menendez-Gonzalez, J.; Iglesias, J.M.; Allen, L.; et al. Hif-1α and Hif-2α synergize to suppress AML development but are dispensable for disease maintenance. J. Exp. Med. 2015, 212, 2223–2234. [Google Scholar] [CrossRef]
- Chertow, G.M.; Pergola, P.E.; Farag, Y.M.; Agarwal, R.; Arnold, S.; Bako, G.; Block, G.A.; Burke, S.; Castillo, F.P.; Jardine, A.G.; et al. Vadadustat in Patients with Anemia and Non-Dialysis-Dependent CKD. N. Engl. J. Med. 2021, 384, 1589–1600. [Google Scholar] [CrossRef]
- Eckardt, K.-U.; Agarwal, R.; Aswad, A.; Awad, A.; Block, G.A.; Bacci, M.R.; Farag, Y.M.; Fishbane, S.; Hubert, H.; Jardine, A.; et al. Safety and Efficacy of Vadadustat for Anemia in Patients Undergoing Dialysis. N. Engl. J. Med. 2021, 384, 1601–1612. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, L.; Wei, X.; Long, M.; Du, Y. Roxadustat: Not just for anemia. Front. Pharmacol. 2022, 13, 971795. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Liu, B.-C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Warfel, N.A. Defining the mechanisms underlying cyclin dependent kinase control of HIF-1α. Oncotarget 2022, 13, 454–455. [Google Scholar] [CrossRef]
- Flamme, I.; Oehme, F.; Ellinghaus, P.; Jeske, M.; Keldenich, J.; Thuss, U. Mimicking hypoxia to treat anemia: HIF-stabilizer BAY 85-3934 (Molidustat) stimulates erythropoietin pro-duction without hypertensive effects. PLoS ONE 2014, 9, e111838. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Eckardt, K.U. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol. 2016, 12, 157–168. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heck-Swain, K.-L.; Koeppen, M. The Intriguing Role of Hypoxia-Inducible Factor in Myocardial Ischemia and Reperfusion: A Comprehensive Review. J. Cardiovasc. Dev. Dis. 2023, 10, 215. https://doi.org/10.3390/jcdd10050215
Heck-Swain K-L, Koeppen M. The Intriguing Role of Hypoxia-Inducible Factor in Myocardial Ischemia and Reperfusion: A Comprehensive Review. Journal of Cardiovascular Development and Disease. 2023; 10(5):215. https://doi.org/10.3390/jcdd10050215
Chicago/Turabian StyleHeck-Swain, Ka-Lin, and Michael Koeppen. 2023. "The Intriguing Role of Hypoxia-Inducible Factor in Myocardial Ischemia and Reperfusion: A Comprehensive Review" Journal of Cardiovascular Development and Disease 10, no. 5: 215. https://doi.org/10.3390/jcdd10050215