
Lipoprotein(a): Its Association with Calcific Aortic Valve Stenosis, the Emerging RNA-Related Treatments and the Hope for a New Era in “Treating” Aortic Valve Calcification

Abstract
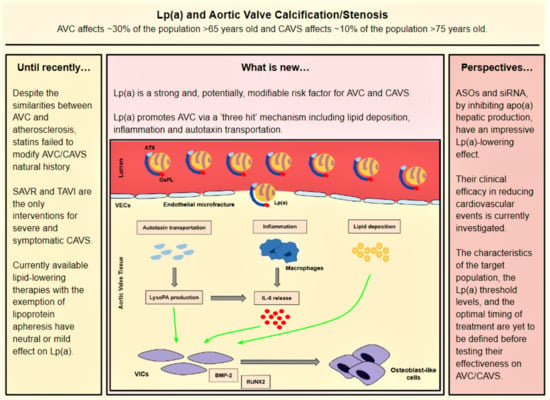
:
1. Introduction
2. Association of Lp(a) with Aortic Stenosis
2.1. Lp(a) and AVC in the General Population
2.2. Lp(a), AVC/CAVS in Patients with Heterozygous Familial Hypercholesterolemia
2.3. Lp(a), AVC/CAVS in Patients with Bicuspid Aortic Valve
2.4. Lp(a) and AVC/CAVS; Causation
2.5. Lp(a) and Progression of AVC/CAVS; an Association in Question
2.6. Pathophysiology
- (a)
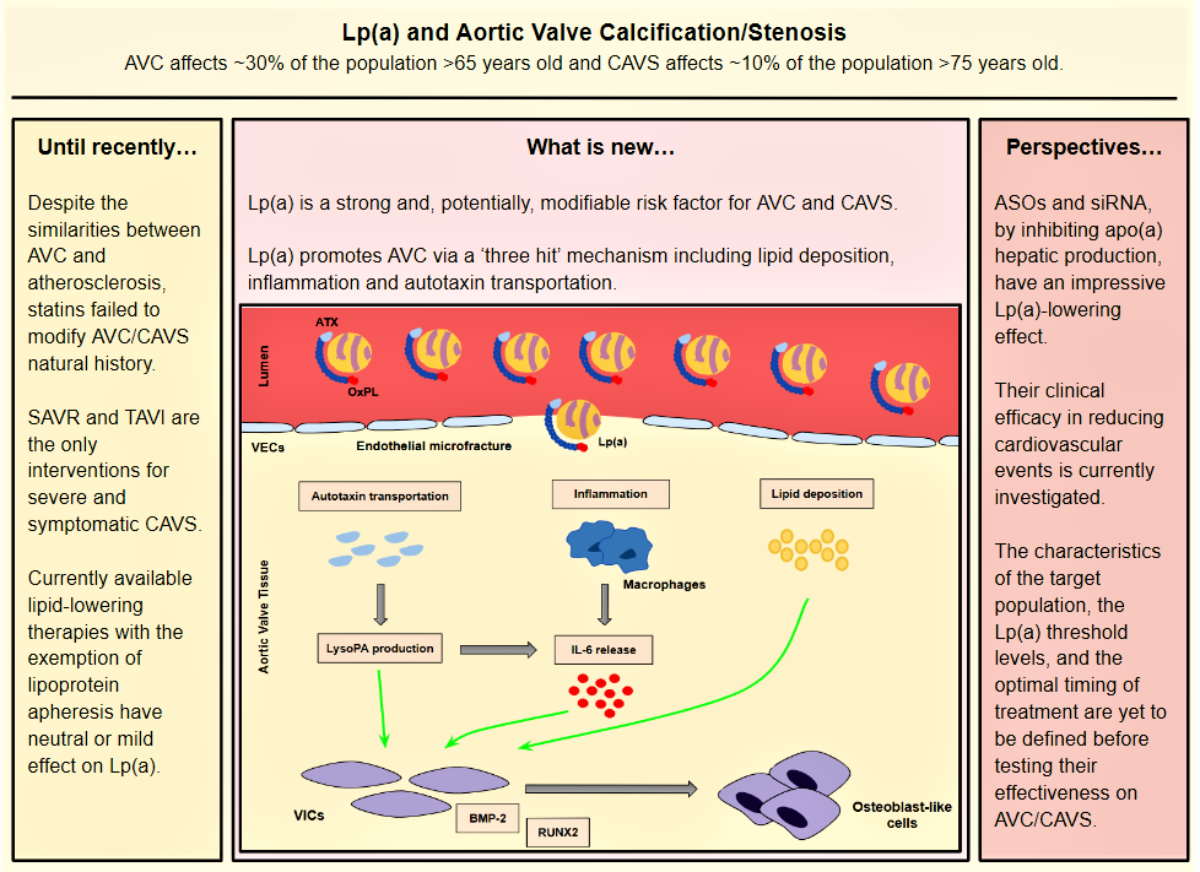
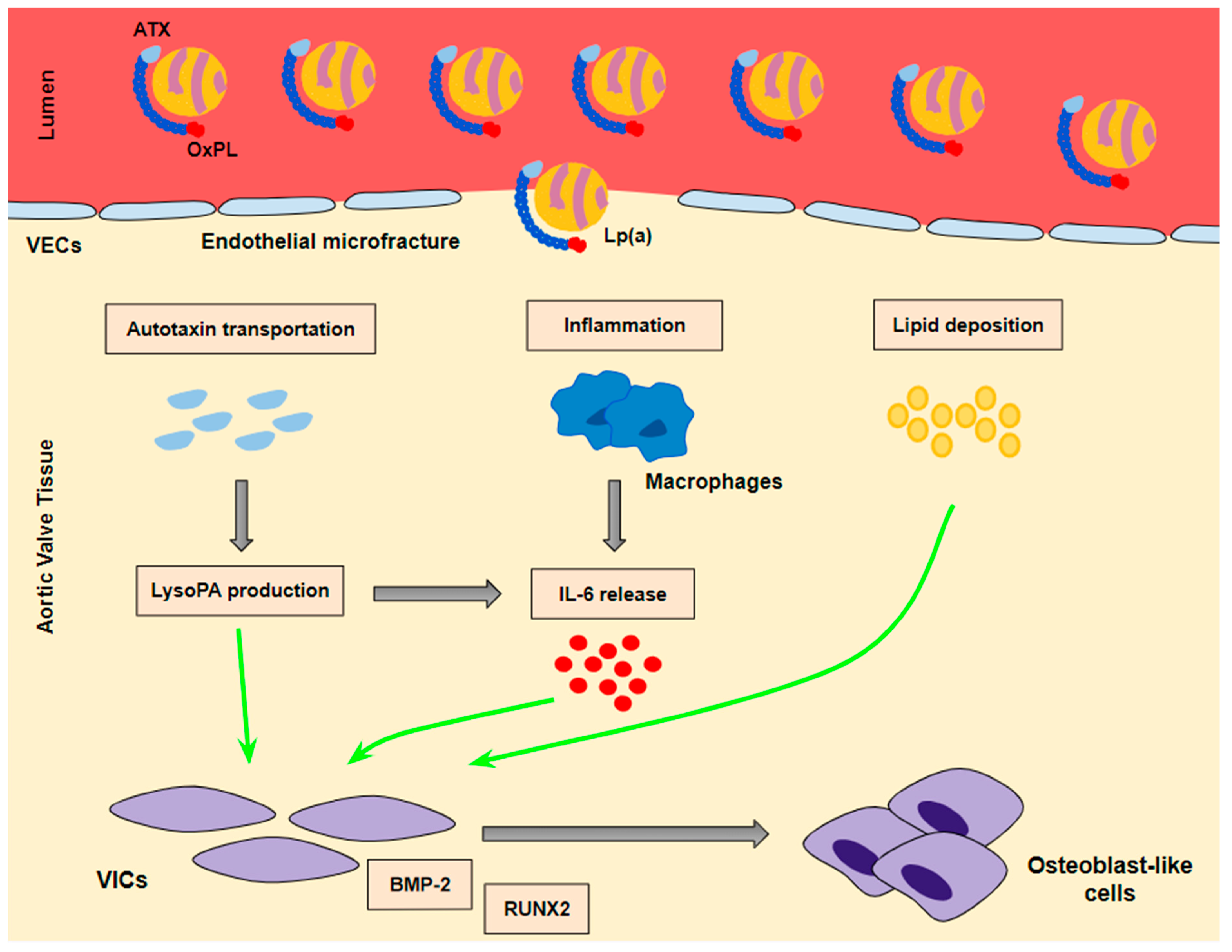
- Lp(a) still preserves LDL properties and, thus, significantly contributes to intravalvular lipid deposition. Some studies propose that Lp(a) also has a role in the wound-healing process. Given the mechanical stress to which aortic valve leaflets are continuously submitted and the subsequent microfractures in their structure, Lp(a) may have an essential role in the lipid deposition process conducted via the healing process. Lipid deposition seems to contribute to VIC differentiation via the BMP2 pathway activation [34].
- (b)
- The OxPL content of apo(a) and its pro-inflammatory properties seem to be key factors of AVC. Inflammation is believed to lead to the activation of the calcification process via the activation of an innate immune response. OxPLs exhibit damage-associated molecular patterns (DAMPs) which, through toll-like receptors (TLRs) expressed on the VIC surface and the Nuclear factor kappa B (NF-kB) pathway, lead to the expression of IL-6. In vitro studies have demonstrated that IL-6 has the potential to activate the BMP2 pathway, thus leading to VIC differentiation [34].
- (c)
- Autotaxin, an oxidizing enzyme, may also have a crucial role in VIC transition. By binding to the Lp(a) molecule, autotaxin is transferred to aortic valve leaflets and triggers the oxidative transformation of phospholipid lysophosphatidylcholine (LysoPC) to lysophosphatidic acid (LysoPA). LysoPA promotes the expression of IL-6 and the activation of the BMP2 pathway leading, once again, to VIC differentiation into osteoblast-like cells, and finally to AVC [34,35].
3. The Failure of Statins to Affect the Progression of AS
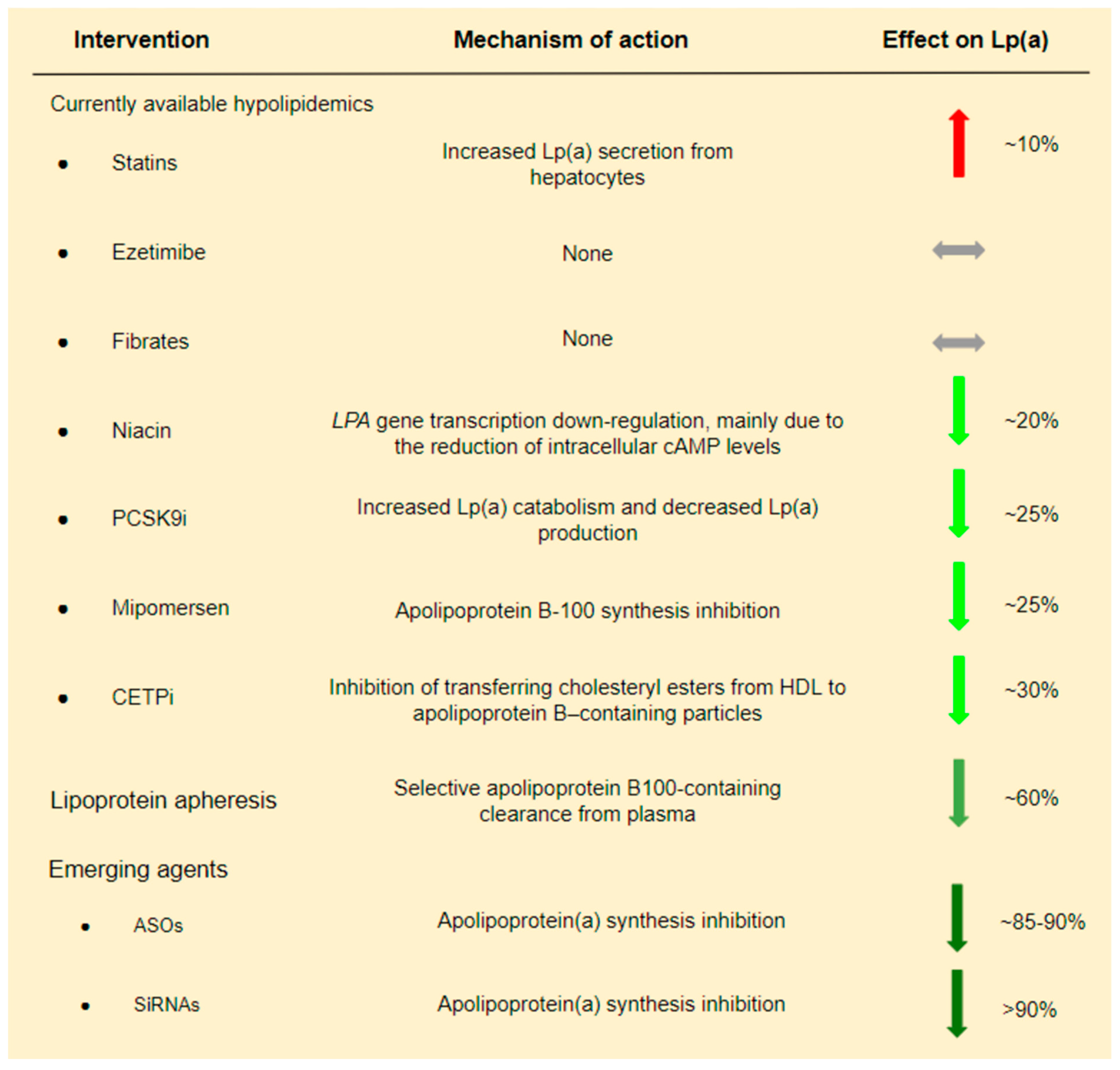
4. Currently Available Lipid-Lowering Therapies and Their Impact on Lp(a)
5. Emerging Treatments
6. Novel Lp(a)-Lowering Therapies: The Dawn of Pharmaceutical Treatment of AS?
- (1)
- What is the best technique to assess the effectiveness of Lp(a)-lowering agents? Echocardiography is an excellent tool to assess AVS, but it is unable to quantitate AVC. On the contrary, CT can measure AVC and can objectively evaluate the effectiveness of the Lp(a)-lowering treatments which will possibly exert their beneficial effect by delaying the calcification process.
- (2)
- Lp(a)-lowering agents should be ideally tested after the completion of phase III trials which explore their safety and effectiveness on cardiovascular events.
- (3)
- How early should Lp(a)-lowering agents be given? “The earlier, the better” principle of statins administration in acute coronary syndrome is likely to be applicable in the setting of AVC. There is data suggesting that the impact of Lp(a) on the progression of calcification may be weak once AVC has been initiated [27].
- (4)
- What is the target population that is more likely to benefit from the new pharmaceutical interventions? It is appropriate to test the new Lp(a)-lowering therapies in populations at high risk of developing AVC/CAVS, i.e., patients with high Lp(a) levels and BAV, or possibly heFH patients? Particularly, in the case of heFH, by lowering Lp(a) it is possible to obtain a dual beneficial effect, i.e., a reduction of cardiovascular events and the prevention of AVC/CAVS.
- (5)
- What will be the threshold Lp(a) levels which will justify a pharmaceutical intervention? In the Lp(a)HORIZON trial, pelacarsen is tested in patients with established CVD and Lp(a) ≥ 70 mg/dL. For the prevention of AVC/CAVS it might be sensible to set higher Lp(a) cutoff levels.
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASOs | Antisense oligonucleotides |
| AS | Aortic Stenosis |
| ATX | Autotaxin |
| AVC | Aortic Valve Calcification |
| BMP2 | Bone morphogenetic protein-2 |
| CAVS | Calcific Aortic Valve Stenosis |
| CETP | Cholesteryl ester transfer protein |
| CT | Computed Tomography |
| CVD | Cardiovascular Disease |
| HDL | High density lipoprotein |
| IL-6 | Interleukin-6 |
| K | Kringle domain |
| LDL | Low-density lipoprotein |
| Lp(a) | Lipoprotein(a) |
| LysoPA | Lysophosphatidic acid |
| OxPL | Oxidized phospholipids |
| PCSK9i | Proprotein convertase subtilisin/kexin type 9 inhibitors |
| RUNX2 | Runt-related transcription factor 2 |
| siRNAs | Small interfering RNAs |
| VECs | Valve endothelial cells |
| VICs | Valve interstitial cells |
References
- Osnabrugge, R.; Mylotte, D.; Head, S.; Van Mieghem, N.; Nkomo, V.; LeReun, C.; Bogers, J.J.C.A.; Piazza, N.; Kappetein, A.P. Aortic Stenosis in the Elderly. J. Am. Coll. Cardiol. 2013, 62, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Jander, N.; Minners, J.; Holme, I.; Gerdts, E.; Boman, K.; Brudi, P.; Chambers, B.J.; Egstrup, K.; Kesaniemi, Y.A.; Malbecq, W.; et al. Outcome of Patients With Low-Gradient “Severe” Aortic Stenosis and Preserved Ejection Fraction. Circulation 2011, 123, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, F.; Mora, S.; Stroes, E.; Ference, B.; Arsenault, B.; Berglund, L.; Dweck, R.M.; Koschinsky, M.; Gilles Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and Aortic Stenosis: A European atherosclerosis society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
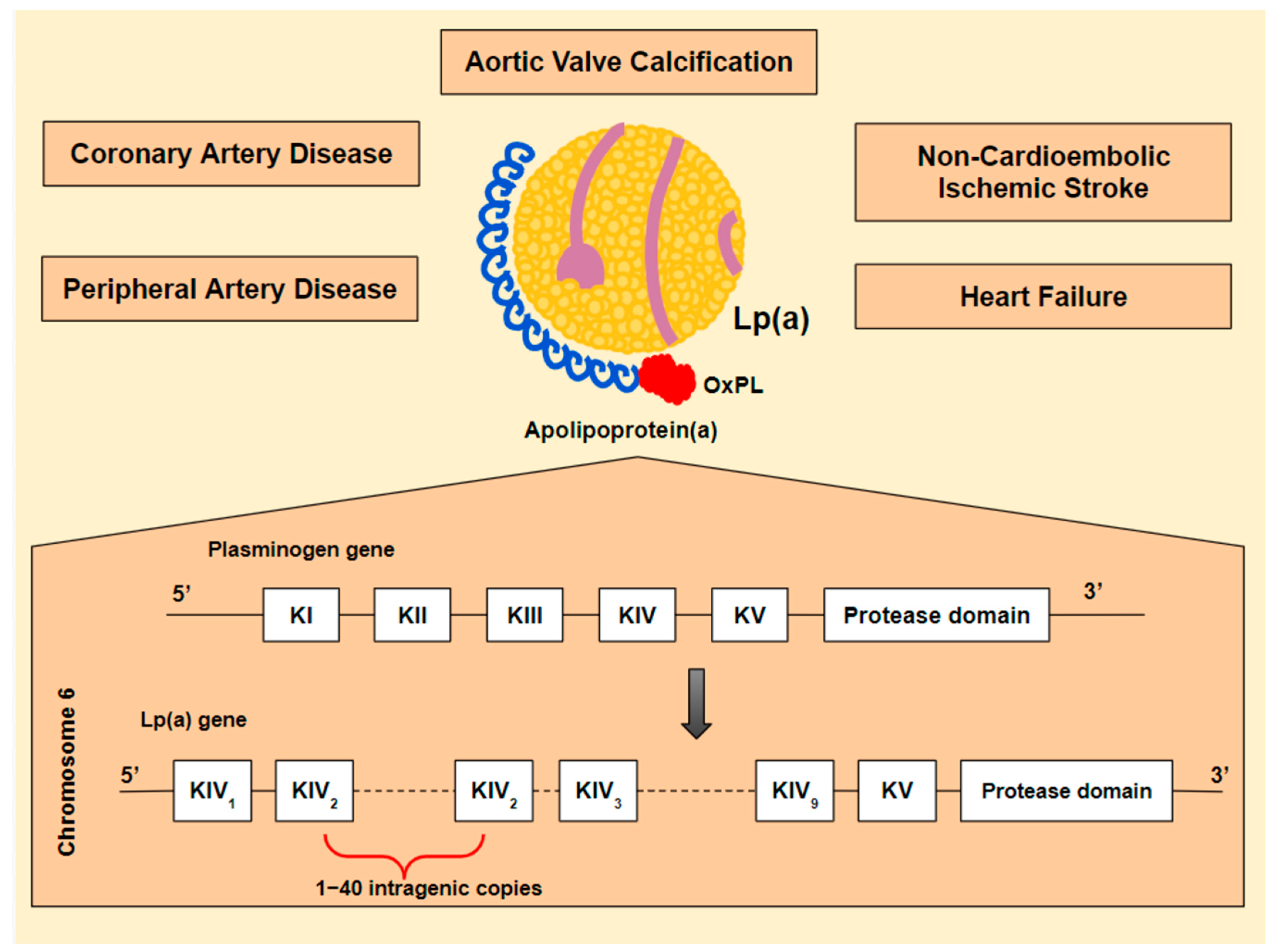
- Gurdasani, D.; Sjouke, B.; Tsimikas, S.; Hovingh, G.; Luben, R.; Wainwright, N.; Pomilla, C.; Wareham, J.N.; Khaw, K.-T.; Boekholdt, M.S.; et al. Lipoprotein(a) and Risk of Coronary, Cerebrovascular, and Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 3058–3065. [Google Scholar] [CrossRef] [Green Version]
- Erqou, S.; Kaptoge, S.; Perry, P.; Di Angelantonio, E.; Thompson, A.; White, I. Lipoprotein(a) Concentration and the Risk of Coronary Heart Disease, Stroke, and Nonvascular Mortality. JAMA 2009, 302, 412. [Google Scholar]
- Kamstrup, P.; Nordestgaard, B. Elevated Lipoprotein(a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC Heart Fail. 2016, 4, 78–87. [Google Scholar] [CrossRef]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein(a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef] [Green Version]
- Hancock, M.; Boffa, M.; Marcovina, S.; Nesheim, M.; Koschinsky, M. Inhibition of Plasminogen Activation by Lipoprotein(a). J. Biol. Chem. 2003, 278, 23260–23269. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.; Koschinsky, M. Mechanisms of Lipoprotein(a) Pathogenicity. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1550–1551. [Google Scholar] [CrossRef] [Green Version]
- Kamstrup, P.; Tybjærg-Hansen, A.; Nordestgaard, B. Genetic Evidence That Lipoprotein (a) Associates With Atherosclerotic Stenosis Rather Than Venous Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1732–1741. [Google Scholar] [CrossRef] [Green Version]
- Tselepis, A. Oxidized phospholipids and lipoprotein-associated phospholipase A2 as important determinants of Lp(a) functionality and pathophysiological role. J. Biomed. Res. 2016, 32, 13. [Google Scholar]
- Tsimikas, S. A Test in Context: Lipoprotein(a). J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Bergmark, C.; Dewan, A.; Orsoni, A.; Merki, E.; Miller, E.; Shin, M.; Binder, J.C.; Hörkkö, S.; Krauss, M.R.; Chapman, J.M.; et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J. Lipid Res. 2008, 49, 2230–2239. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Chan, K.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.; Tam, W.J.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Hughes, B.; Chahoud, G.; Mehta, J. Aortic stenosis: Is it simply a degenerative process or an active atherosclerotic process? Clin. Cardiol. 2005, 28, 111–114. [Google Scholar] [CrossRef]
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. J. Am. Coll. Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Fedak, P.; Verma, S.; David, T.; Leask, R.; Weisel, R.; Butany, J. Clinical and Pathophysiological Implications of a Bicuspid Aortic Valve. Circulation 2002, 106, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, T.; Kuroda, T.; Yamasawa, M.; Nishinaga, M.; Mitsuhashi, T.; Seino, Y.; Nagoh, N.; Kayaba, K.; Yamada, S.; Matsuo, H.; et al. Correlation between lipoprotein (a) and aortic valve sclerosis assessed by echocardiography (the JMS Cardiac Echo and Cohort Study). Am. J. Cardiol. 1995, 76, 928–932. [Google Scholar] [CrossRef]
- Kamstrup, P.; Tybjærg-Hansen, A.; Nordestgaard, B. Elevated Lipoprotein(a) and Risk of Aortic Valve Stenosis in the General Population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, B.; Boekholdt, S.; Dubé, M.; Rhéaume, É.; Wareham, N.; Khaw, K.; Sandhu, S.M.; Tardif, J.-C. Lipoprotein(a) Levels, Genotype, and Incident Aortic Valve Stenosis. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Rallidis, L.; Naoumova, R.P.; Thompson, G.R.; Nihoyannopoulos, P. Extent and severity of atherosclerotic involvement of the aortic valve and root in familial hypercholesterolaemia. Heart 1998, 80, 583–590. [Google Scholar] [CrossRef]
- Rallidis, L.; Nihoyannopoulos, P.; Thompson, G.R. Aortic stenosis in homozygous familial hypercholesterolaemia. Heart 1996, 76, 84–85. [Google Scholar] [CrossRef] [Green Version]
- Soran, H.; Adam, S.; Iqbal, Z.; Durrington, P. Non-HDL cholesterol should not generally replace LDL cholesterol in the management of hyperlipidaemia. Curr. Opin. Lipidol. 2019, 30, 263–372. [Google Scholar] [CrossRef]
- Vuorio, A.; Watts, G.F.; Kovanen, P.T. Lipoprotein(a) as a risk factor for calcific aortic valvulopathy in heterozygous familial hypercholesterolemia. Atherosclerosis 2019, 281, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Vongpromek, R.; Bos, S.; ten Kate, G.; Yahya, R.; Verhoeven, A.; de Feyter, P.; Kronenberg, F.; van Lennep, J.E.R.; Sijbrands, E.J.G.; Mulder, M.T. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J. Intern. Med. 2015, 278, 166–173. [Google Scholar] [CrossRef]
- Perrot, N.; Thériault, S.; Dina, C.; Chen, H.; Boekholdt, S.; Rigade, S.; Després, A.-A.; Poulin, A.; Capoulade, R.; Le Tourneau, T.; et al. Genetic Variation in LPA, Calcific Aortic Valve Stenosis in Patients Undergoing Cardiac Surgery, and Familial Risk of Aortic Valve Microcalcification. JAMA Cardiol. 2019, 4, 620. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, Y.; Daghem, M.; Tzolos, E.; Meah, M.; Doris, M.; Moss, A.; Kwiecinski, J.; Kroon, J.; Nurmohamed, N.S.; van der Harst, P.; et al. Association of Lipoprotein(a) With Atherosclerotic Plaque Progression. J. Am. Coll. Cardiol. 2022, 79, 223–233. [Google Scholar] [CrossRef]
- Sticchi, E.; Giusti, B.; Cordisco, A.; Gori, A.M.; Sereni, A.; Sofi, F.; Mori, F.; Colonna, S.; Fugazzaro, M.P.; Pepe, G.; et al. Role of lipoprotein(a) and LPA Kiv2 repeat polymorphism in bicuspid aortic valve stenosis and calcification: A proof of concept study. Intern. Emerg. Med. 2018, 14, 45–50. [Google Scholar] [CrossRef]
- Thanassoulis, G.; Campbell, Y.C.; Owens, S.D.; Smith, J.G.; Smith, V.A.; Peloso, M.G.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Yeang, C.; Chan, K.; Pibarot, P.; Tsimikas, S. Association of Mild to Moderate Aortic Valve Stenosis Progression With Higher Lipoprotein(a) and Oxidized Phospholipid Levels. JAMA Cardiol. 2018, 3, 1212. [Google Scholar] [CrossRef]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.; Doris, M.K.; White, A.C.; Timmers, N.K.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef]
- Kaiser, Y.; van der Toorn, E.J.; Singh, S.S.; Zheng, H.K.; Kavousi, M.; Sijbrands, J.G.E.; Stroes, E.S.G.; Vernooij, M.W.; de Rijke, Y.B.; Boekholdt, S.M.; et al. Lipoprotein(a) is associated with the onset but not the progression of aortic valve calcification. Eur. Heart J. 2022, 43, 3960–3967. [Google Scholar] [CrossRef]
- Dweck, R.M.; Khaw, J.H.; Sng GK, Z.; Luo, E.L.C.; Baird, A.; Williams, M.C.; Makiello, P.; Mirsadraee, S.; Joshi, N.V.; van Beek, E.J.R.; et al. Aortic stenosis, atherosclerosis, and skeletal bone: Is there a common link with calcification and inflammation? Eur. Heart J. 2013, 34, 1567–1574. [Google Scholar] [CrossRef] [Green Version]
- Akahori, H.; Tsujino, T.; Masuyama, T.; Ishihara, M. Mechanisms of aortic stenosis. J. Cardiol. 2018, 71, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, P.; Boulanger, M. Autotaxin and Lipoprotein Metabolism in Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2019, 6, 18. [Google Scholar] [CrossRef]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J.; ASTRONOMER Investigators. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Cowell, S.; Newby, D.; Prescott, R.; Bloomfield, P.; Reid, J.; Northridge, D.; Boon, N.A. A Randomized Trial of Intensive Lipid-Lowering Therapy in Calcific Aortic Stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef]
- Lee, S.-E.; Chang, H.-J.; Sung, J.M.; Park, H.-B.; Heo, R.; Rizvi, A.; Lin, F.Y.; Kumar, A.; Hadamitzky, M.; Kim, Y.J.; et al. Effects of statins on coronary atherosclerotic plaques. JACC Cardiovasc. Imaging 2018, 11, 1475–1484. [Google Scholar]
- Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Leebmann, J.; Lehmacher, W.; Kamstrup, P.K.; Nordestgaard, B.G.; et al. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2019–2027. [Google Scholar] [CrossRef] [Green Version]
- Moriarty, P.; Gray, J.; Gorby, L. Lipoprotein apheresis for lipoprotein(a) and cardiovascular disease. J. Clin. Lipidol. 2019, 13, 894–900. [Google Scholar] [CrossRef]
- Schettler, V.; Neumann, C.; Peter, C.; Zimmermann, T.; Julius, U.; Hohenstein, B.; Roeseler, E.; Heigl, F.; Grützmacher, P.; Blume, H.; et al. Lipoprotein apheresis is an optimal therapeutic option to reduce increased Lp(a) levels. Clin. Res. Cardiol. Suppl. 2019, 14 (Suppl. S1), 33–38. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, B.; Julius, U.; Lansberg, P.; Jaeger, B.; Mellwig, K.; Weiss, N.; Graehlert, X.; Roeder, I.; Ramlow, W. Rationale and design of MultiSELECt: A European Multi center Study on the Effect of Lipoprotein(a) Elimination by lipoprotein apheresis on Cardiovascular outcomes. Atheroscler. Suppl. 2017, 30, 180–186. [Google Scholar] [CrossRef]
- Hernández, C.; Francisco, G.; Ciudin, A.; Chacón, P.; Montoro, B.; Llaverias, G.; Blanco-Vaca, F.; Simó, R. Effect of atorvastatin on lipoprotein(a) and interleukin-10: A randomized placebo-controlled trial. Diabetes Metab. 2011, 37, 124–130. [Google Scholar] [CrossRef]
- Gonbert, S.; Malinsky, S.; Sposito, A.; Laouenan, H.; Doucet, C.; Chapman, M.; Thillet, J. Atorvastatin lowers lipoprotein(a) but not apolipoprotein (a) fragment levels in hypercholesterolemic subjects at high cardiovascular risk. Atherosclerosis 2002, 164, 305–311. [Google Scholar] [CrossRef]
- Yahya, R.; Berk, K.; Verhoeven, A.; Bos, S.; van der Zee, L.; Touw, J.; Erhart, G.; Kronenberg, F.; Timman, R.; Sijbrands, E.; et al. Statin treatment increases lipoprotein(a) levels in subjects with low molecular weight apolipoprotein(a) phenotype. Atherosclerosis 2019, 289, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, B.; Petrides, F.; Tabet, F.; Bao, W.; Hovingh, G.; Boekholdt, S.; Ramin-Mangata, S.; Meilhac, O.; DeMicco, D.; Rye, K.-A.; et al. Effect of atorvastatin, cholesterol ester transfer protein inhibition, and diabetes mellitus on circulating proprotein subtilisin kexin type 9 and lipoprotein(a) levels in patients at high cardiovascular risk. J. Clin. Lipidol. 2018, 12, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Khera, A.; Everett, B.; Caulfield, M.; Hantash, F.; Wohlgemuth, J.; Ridker, P.; Mora, S. Lipoprotein(a) Concentrations, Rosuvastatin Therapy, and Residual Vascular Risk. Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Gordts, P.; Nora, C.; Yeang, C.; Witztum, J. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2019, 41, 2275–2284. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Ju, J.; Fan, Y.; Xu, H. Effect of different types and dosages of statins on plasma lipoprotein(a) levels: A network meta-analysis. Pharmacol. Res. 2021, 163, 105275. [Google Scholar] [CrossRef]
- Farmakis, I.; Doundoulakis, I.; Pagiantza, A.; Zafeiropoulos, S.; Antza, C.; Karvounis, H.; Giannakoulas, G. Lipoprotein(a) Reduction With Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors: A Systematic Review and Meta-analysis. J. Cardiovasc. Pharmacol. 2021, 77, 397–407. [Google Scholar] [CrossRef]
- Mahmood, T.; Minnier, J.; Ito, M.; Li, Q.; Koren, A.; Kam, I.; Fazio, S.; Shapiro, M.D. Discordant responses of plasma low-density lipoprotein cholesterol and lipoprotein(a) to alirocumab: A pooled analysis from 10 ODYSSEY Phase 3 studies. Eur. J. Prev. Cardiol. 2020, 28, 816–822. [Google Scholar] [CrossRef] [Green Version]
- Croyal, M.; Tran, T.; Blanchard, R.; Le Bail, J.; Villard, E.; Poirier, B.; Aguesse, A.; Billon-Crossouard, S.; Ramin-Mangata, S.; Blanchard, V.; et al. PCSK9 inhibition with alirocumab reduces lipoprotein(a) levels in nonhuman primates by lowering apolipoprotein (a) production rate. Clin. Sci. 2018, 132, 1075–1083. [Google Scholar] [CrossRef]
- Watts, G.; Chan, D.; Pang, J.; Ma, L.; Ying, Q.; Aggarwal, S.; Marcovina, S.M.; Barrett, P.H.R. PCSK9 Inhibition with alirocumab increases the catabolism of lipoprotein(a) particles in statin-treated patients with elevated lipoprotein(a). Metabolism 2020, 107, 154221. [Google Scholar] [CrossRef]
- Watts, G.; Chan, D.; Somaratne, R.; Wasserman, S.; Scott, R.; Marcovina, S.; Barrett, P.H.R. Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur. Heart J. 2018, 39, 2577–2585. [Google Scholar] [CrossRef]
- Sahebkar, A.; Reiner, Ž.; Simental-Mendía, L.; Ferretti, G.; Cicero, A. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism 2016, 65, 1664–1678. [Google Scholar] [CrossRef]
- Nicholls, S.; Ruotolo, G.; Brewer, H.; Wang, M.; Liu, L.; Willey, M.; Deeg, M.A.; Krueger, K.A.; Nissen, S.E. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 519–527.e4. [Google Scholar] [CrossRef]
- Aim-High Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [Green Version]
- Lincoff, A.M.; Nicholls, J.S.; Riesmeyer, S.J.; Barter, H.P.; Brewe, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Pineda, A.L.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- Burgess, S.; Ference, B.; Staley, J.; Freitag, D.; Mason, A.; Nielsen, S.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies. JAMA Cardiol. 2018, 3, 619. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.; Witztum, J.; Bennett, C.; Baker, B. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.; Raal, F.; Catapano, A.; Witztum, J.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an Antisense Oligonucleotide to Apolipoprotein B-100, Reduces Lipoprotein(a) in Various Populations With Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, E.; Vogt, A.; Crispin, A.; Altenhofer, J.; Riks, I.; Parhofer, K. Effect of mipomersen on LDL-cholesterol in patients with severe LDL-hypercholesterolaemia and atherosclerosis treated by lipoprotein apheresis (The MICA-Study). Atherosclerosis 2017, 259, 20–25. [Google Scholar] [CrossRef]
- Merki, E.; Graham, M.; Mullick, A.; Miller, E.; Crooke, R.; Pitas, R.; Witztim, J.L.; Tsimikas, S. Antisense Oligonucleotide Directed to Human Apolipoprotein B-100 Reduces Lipoprotein (a) Levels and Oxidized Phospholipids on Human Apolipoprotein B-100 Particles in Lipoprotein(a) Transgenic Mice. Circulation 2008, 118, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Merki, E.; Graham, M.; Taleb, A.; Leibundgut, G.; Yang, X.; Miller, E.; Fu, W.; Mullick, A.E.; Lee, R.; Willeit, P.; et al. Antisense Oligonucleotide Lowers Plasma Levels of Apolipoprotein(a) and Lipoprotein(a) in Transgenic Mice. J. Am. Coll. Cardiol. 2011, 57, 1611–1621. [Google Scholar] [CrossRef] [Green Version]
- Graham, M.; Viney, N.; Crooke, R.; Tsimikas, S. Antisense inhibition of apolipoprotein(a) to lower plasma lipoprotein(a) levels in humans. J. Lipid Res. 2016, 57, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Viney, N.; Hughes, S.; Singleton, W.; Graham, M.; Baker, B.; Burkey, J.L.; Yang, O.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Viney, N.; van Capelleveen, J.; Geary, R.; Xia, S.; Tami, J.; Yu, R.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein (a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Tsimikas, S.; Moriarty, P.; Stroes, E. Emerging RNA Therapeutics to Lower Blood Levels of Lp(a). J. Am. Coll. Cardiol. 2021, 77, 1576–1589. [Google Scholar] [CrossRef]
- Assessing the Impact of Lipoprotein(a) Lowering With Pelacarsen (TQJ230) on Major Cardiovascular Events in Patients with CVD (Lp(a)HORIZON). NCT Number; NCT04023552. Available online: https://clinicaltrials.gov/ct2/show/NCT04023552 (accessed on 22 February 2023).
- Tadin-Strapps, M.; Robinson, M.; Le Voci, L.; Andrews, L.; Yendluri, S.; Williams, S.; Bartz, S.; Johns, D.G. Development of Lipoprotein(a) siRNAs for Mechanism of Action Studies in Non-Human Primate Models of Atherosclerosis. J. Cardiovasc. Transl. Res. 2015, 8, 44–53. [Google Scholar] [CrossRef]
- Nissen, S.; Wolski, K.; Balog, C.; Swerdlow, D.; Scrimgeour, A.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals With Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679. [Google Scholar] [CrossRef]
- Evaluate SLN360 in Participants With Elevated Lipoprotein(a) at High Risk of Atherosclerotic Cardiovascular Disease Events, NCT Number; NCT05537571. Available online: https://www.clinicaltrials.gov/ct2/show/NCT05537571?cond=05537571&draw=2&rank=1 (accessed on 22 February 2023).
- Koren, M.; Moriarty, P.; Baum, S.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Stacey Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small interfering RNA to reduce lipoprotein(a) in cardiovascular disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Olpasiran Trials of Cardiovascular Events and Lipoprotein(a) Reduction (OCEAN(a))—Outcomes Trial NCT Number; NCT05581303. Available online: https://clinicaltrials.gov/ct2/show/study/NCT05581303 (accessed on 22 February 2023).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | NCT | Intervention | Participants | Study Type | Primary Endpoint | Results | Status |
|---|---|---|---|---|---|---|---|
| IONIS-APO(a)Rx Type of gene silencing: ASO | 02160899 | SC administration in multiple doses and intervals | 64 patients Cohort A: Lp(a) ≥ 50 and <175 mg/dL Cohort B: Lp(a) ≥ 175 mg/dL | Randomize, Double Blind, Placebo-Controlled, Dose Titration, Phase II Trial | 1. Percent Lp(a) reduction (at days 85 and 99) 2. Number of TEAEs (at 32 weeks) | 1. Lp(a) reduction up 71.6% 2. None TEAE reported | Completed in November 2015 |
| IONIS APO(a)-LRx (AKCEA-APO(a)-LRx, TQJ230, Pelacarsen) Type of gene silencing: ASO | 04023552 (Lp(a) HORIZON-trial) | 80 mg SC administration monthly | 8324 patients between 18 and 80 years old with Lp(a) > 70 mg/dL and established ASCVD | Randomized Double-blind, Placebo-controlled, Multicenter, Phase III Trial | Time to first occurrence of clinical endpoint committee confirmed expanded MACE | Not yet available [In phase II study; Lp(a) reduction up to 92% with 40 mg SC in ascending-doses] | Expected to be completed in May 2025 |
| SLN360 Type of gene silencing: SiRNA | 05537571 | A single SC injection in multiple doses | 160 patients between 18 and 80 years old at high risk of ASCVD events and Lp(a) ≥125 nmol/L | Multi-centre, Randomised, Double-blind, Placebo-controlled, Phase II Study | Time averaged change in Lp(a) from baseline at 36 weeks | Not yet available [In phase I study; Lp(a) reduction up to 98% with 600 mg single dose SC injection. Reduction highly maintained on day 150] | Expected to be completed in November 2024 |
| Olpasiran (AMG 890) Type of gene silencing: SiRNA | 05581303 (OCEAN(a)—Outcomes Trial) | SC injection once Q12W | 6000 patients between 18 and 85 years old with established ASCVD and Lp(a) ≥200 nmol/L during screening | Double-blind, Randomized, Placebo-controlled, Multicenter Phase III Study | Time to CHD death, myocardial infarction, or urgent coronary revascularization, whichever occurs first | Not yet available [In phase II study; Lp(a) reduction up to 101.1% with the 225-mg dose administered every 12 weeks (placebo-adjusted mean percent changes)] | Expected to be completed in December 2026 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsamoulis, D.; Siountri, I.; Rallidis, L.S. Lipoprotein(a): Its Association with Calcific Aortic Valve Stenosis, the Emerging RNA-Related Treatments and the Hope for a New Era in “Treating” Aortic Valve Calcification. J. Cardiovasc. Dev. Dis. 2023, 10, 96. https://doi.org/10.3390/jcdd10030096
Tsamoulis D, Siountri I, Rallidis LS. Lipoprotein(a): Its Association with Calcific Aortic Valve Stenosis, the Emerging RNA-Related Treatments and the Hope for a New Era in “Treating” Aortic Valve Calcification. Journal of Cardiovascular Development and Disease. 2023; 10(3):96. https://doi.org/10.3390/jcdd10030096
Chicago/Turabian StyleTsamoulis, Donatos, Iliana Siountri, and Loukianos S. Rallidis. 2023. "Lipoprotein(a): Its Association with Calcific Aortic Valve Stenosis, the Emerging RNA-Related Treatments and the Hope for a New Era in “Treating” Aortic Valve Calcification" Journal of Cardiovascular Development and Disease 10, no. 3: 96. https://doi.org/10.3390/jcdd10030096