
Characterization of the Blood Microbiome and Comparison with the Fecal Microbiome in Healthy Dogs and Dogs with Gastrointestinal Disease

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Housing
2.2. Fecal and Blood Microbial DNA Extraction and Sequencing
2.3. Computational and Statistical Analysis
3. Results
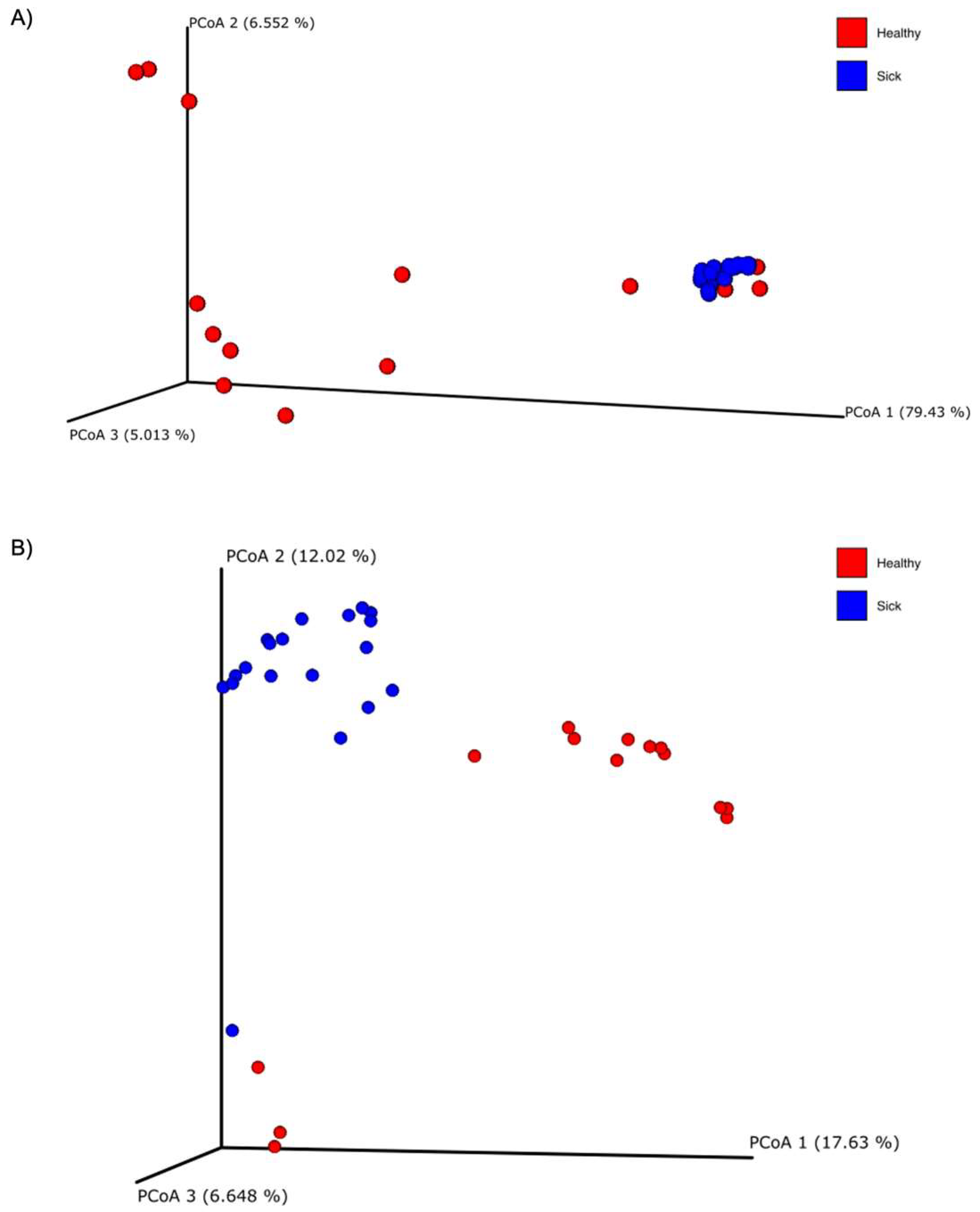
3.1. Translocation of Bacteria from the Gastrointestinal Tract to the Blood
3.2. Characterization of the Blood Microbiome Related to the Gastrointestinal Health Status
3.3. Characterization of the Fecal Microbiome Related to the Gastrointestinal Health Status
3.4. Characterization of Predicted Functionality on Gut and Blood Microbiome in Healthy and Sick Dogs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pilla, R.; Suchodolski, J.S. The Role of the Canine Gut Microbiome and Metabolome in Health and Gastrointestinal Disease. Front. Vet. Sci. 2020, 6, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghose, C.; Ly, M.; Schwanemann, L.K.; Shin, J.H.; Atab, K.; Barr, J.J.; Little, M.; Schooley, R.T.; Chopyk, J.; Pride, D.T. The Virome of Cerebrospinal Fluid: Viruses Where We Once Thought There Were None. Front. Microbiol. 2019, 10, 02061. [Google Scholar] [CrossRef]
- Lathe, R.; St. Clair, D. From Conifers to Cognition: Microbes, Brain and Behavior. Genes Brain Behav. 2020, 19, e12680. [Google Scholar] [CrossRef]
- Kang, Y.; Ji, X.; Guo, L.; Xia, H.; Yang, X.; Xie, Z.; Shi, X.; Wu, R.; Feng, D.; Wang, C.; et al. Cerebrospinal Fluid from Healthy Pregnant Women Does Not Harbor a Detectable Microbial Community. Microbiol. Spectr. 2021, 9, e00769-21. [Google Scholar] [CrossRef]
- Liao, H.; Zhang, Y.; Guo, W.; Wang, X.; Wang, H.; Ye, H.; Wu, K.; Zhang, Y.-H.; Guo, L.; Zhu, Y.; et al. Characterization of the Blood and Cerebrospinal Fluid Microbiome in Children with Bacterial Meningitis and Its Potential Correlation with Inflammation. mSystems 2021, 6, e00049-21. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, G.G.; Amici, D.; Paparelli, M. Incorporation of Nucleosides and Amino-Acids in Human Erythrocyte Suspensions: Possible Relation with a Diffuse Infection of Mycoplasms or Bacteria in the L Form. Nature 1969, 222, 1285–1286. [Google Scholar] [CrossRef]
- Domingue, G.J.; Schlegel, J.U. Novel Bacterial Structures in Human Blood: Cultural Isolation. Infect. Immun. 1977, 15, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Nikkari, S.; McLaughlin, I.J.; Bi, W.; Dodge, D.E.; Relman, D.A. Does Blood of Healthy Subjects Contain Bacterial Ribosomal DNA? J. Clin. Microbiol. 2001, 39, 1956–1959. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, K.; Ando, C.; Tashiro, K.; Kuhara, S.; Okamura, S.; Nakano, S.; Takagi, Y.; Miki, T.; Nakashima, Y.; Hirakawa, H. Polymerase Chain Reaction Detection of Bacterial 16S RRNA Gene in Human Blood. Microbiol. Immunol. 2008, 52, 375–382. [Google Scholar] [CrossRef]
- Païssé, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive Description of Blood Microbiome from Healthy Donors Assessed by 16S Targeted Metagenomic Sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Panaiotov, S.; Filevski, G.; Equestre, M.; Nikolova, E.; Kalfin, R. Cultural Isolation and Characteristics of the Blood Microbiome of Healthy Individuals. Adv. Microbiol. 2018, 8, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-Method Characterization of the Human Circulating Microbiome. Front. Microbiol. 2019, 9, 3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velmurugan, G.; Dinakaran, V.; Rajendhran, J.; Swaminathan, K. Blood Microbiota and Circulating Microbial Metabolites in Diabetes and Cardiovascular Disease. Trends Endocrinol. Metab. 2020, 31, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.K.; Jiang, T.; Al-Rubaye, A.A.; Rhoads, D.D.; Wideman, R.F.; Zhao, J.; Pevzner, I.; Kwon, Y.M. An Investigation into Blood Microbiota and Its Potential Association with Bacterial Chondronecrosis with Osteomyelitis (BCO) in Broilers. Sci. Rep. 2016, 6, 25882. [Google Scholar] [CrossRef] [Green Version]
- Vientós-Plotts, A.I.; Ericsson, A.C.; Rindt, H.; Grobman, M.E.; Graham, A.; Bishop, K.; Cohn, L.A.; Reinero, C.R. Dynamic Changes of the Respiratory Microbiota and Its Relationship to Fecal and Blood Microbiota in Healthy Young Cats. PLoS ONE 2017, 12, e0173818. [Google Scholar] [CrossRef]
- Scarsella, E.; Sandri, M.; Monego, S.D.; Licastro, D.; Stefanon, B. Blood Microbiome: A New Marker of Gut Microbial Population in Dogs? Vet. Sci. 2020, 7, 198. [Google Scholar] [CrossRef]
- Scarsella, E.; Zecconi, A.; Cintio, M.; Stefanon, B. Characterization of Microbiome on Feces, Blood and Milk in Dairy Cows with Different Milk Leucocyte Pattern. Animals 2021, 11, 1463. [Google Scholar] [CrossRef]
- Tilahun, Y.; Pinango, J.Q.; Johnson, F.; Lett, C.; Smith, K.; Gipson, T.; McCallum, M.; Hoyt, P.; Tritt, A.; Yadav, A.; et al. Transcript and Blood-Microbiome Analysis towards a Blood Diagnostic Tool for Goats Affected by Haemonchus Contortus. Sci. Rep. 2022, 12, 5362. [Google Scholar] [CrossRef]
- Iwai, T. Periodontal Bacteremia and Various Vascular Diseases. J. Periodontal Res. 2009, 44, 689–694. [Google Scholar] [CrossRef]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological Bacterial Translocation in Liver Cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-Mediated Dendritic Cell Access to the Intestinal Lumen and Bacterial Clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Castillo, D.J.; Rifkin, R.F.; Cowan, D.A.; Potgieter, M. The Healthy Human Blood Microbiome: Fact or Fiction? Front. Cell. Infect. Microbiol. 2019, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Deng, P.; Swanson, K.S. Gut Microbiota of Humans, Dogs and Cats: Current Knowledge and Future Opportunities and Challenges. Br. J. Nutr. 2015, 113, S6–S17. [Google Scholar] [CrossRef] [PubMed]
- Forster, G.M.; Stockman, J.; Noyes, N.; Heuberger, A.L.; Broeckling, C.D.; Bantle, C.M.; Ryan, E.P. A Comparative Study of Serum Biochemistry, Metabolome and Microbiome Parameters of Clinically Healthy, Normal Weight, Overweight, and Obese Companion Dogs. Top. Companion Anim. Med. 2018, 33, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazcorro, J.F.; Barcenas-Walls, J.R.; Suchodolski, J.S.; Steiner, J.M. Molecular Assessment of the Fecal Microbiota in Healthy Cats and Dogs before and during Supplementation with Fructo-Oligosaccharides (FOS) and Inulin Using High-Throughput 454-Pyrosequencing. PeerJ 2017, 5, e3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilson, Å.; Ramadan, Z.; Li, Q.; Hedhammar, Å.; Reynolds, A.; Spears, J.; Labuda, J.; Pelker, R.; Björkstén, B.; Dicksved, J.; et al. Disentangling Factors That Shape the Gut Microbiota in German Shepherd Dogs. PLoS ONE 2018, 13, e0193507. [Google Scholar] [CrossRef]
- Cintio, M.; Scarsella, E.; Sgorlon, S.; Sandri, M.; Stefanon, B. Gut Microbiome of Healthy and Arthritic Dogs. Vet. Sci. 2020, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Coelho, L.P.; Kultima, J.R.; Costea, P.I.; Fournier, C.; Pan, Y.; Czarnecki-Maulden, G.; Hayward, M.R.; Forslund, S.K.; Schmidt, T.S.B.; Descombes, P.; et al. Similarity of the Dog and Human Gut Microbiomes in Gene Content and Response to Diet. Microbiome 2018, 6, 72. [Google Scholar] [CrossRef]
- Allenspach, K.; Mochel, J.P. Current Diagnostics for Chronic Enteropathies in Dogs. Vet. Clin. Pathol. 2022, 50, 18–28. [Google Scholar] [CrossRef]
- Simpson, K.W.; Jergens, A.E. Pitfalls and Progress in the Diagnosis and Management of Canine Inflammatory Bowel Disease. Vet. Clin. Small Anim. Pract. 2011, 41, 381–398. [Google Scholar] [CrossRef]
- Packey, C.D.; Sartor, R.B. Commensal Bacteria, Traditional and Opportunistic Pathogens, Dysbiosis and Bacterial Killing in Inflammatory Bowel Diseases. Curr. Opin. Infect. Dis. 2009, 22, 292. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.; Round, J.L. Defining Dysbiosis and Its Influence on Host Immunity and Disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An Effective Distance Metric for Microbial Community Comparison. ISME J 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J. A New Method for Non-Parametric Multivariate Analysis of Variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Ridyard, A.E.; Brown, J.K.; Rhind, S.M.; Else, R.W.; Simpson, J.W.; Miller, H.R.P. Apical Junction Complex Protein Expression in the Canine Colon: Differential Expression of Claudin-2 in the Colonic Mucosa in Dogs With Idiopathic Colitis. J. Histochem. Cytochem. 2007, 55, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Suchodolski, J.S. Diagnosis and Interpretation of Intestinal Dysbiosis in Dogs and Cats. Vet. J. 2016, 215, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.S.; Pratt-Phillips, S.; Gonzalez, L.M. Alterations in Intestinal Permeability: The Role of the “Leaky Gut” in Health and Disease. J. Equine Vet. Sci. 2017, 52, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Tizard, I.R.; Jones, S.W. The Microbiota Regulates Immunity and Immunologic Diseases in Dogs and Cats. Vet. Clin. Small Anim. Pract. 2018, 48, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Suchodolski, J.S.; Markel, M.E.; Garcia-Mazcorro, J.F.; Unterer, S.; Heilmann, R.M.; Dowd, S.E.; Kachroo, P.; Ivanov, I.; Minamoto, Y.; Dillman, E.M.; et al. The Fecal Microbiome in Dogs with Acute Diarrhea and Idiopathic Inflammatory Bowel Disease. PLoS ONE 2012, 7, e51907. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Baeza, Y.; Hyde, E.R.; Suchodolski, J.S.; Knight, R. Dog and Human Inflammatory Bowel Disease Rely on Overlapping yet Distinct Dysbiosis Networks. Nat. Microbiol. 2016, 1, 16177. [Google Scholar] [CrossRef]
- Isaiah, A.; Parambeth, J.C.; Steiner, J.M.; Lidbury, J.A.; Suchodolski, J.S. The Fecal Microbiome of Dogs with Exocrine Pancreatic Insufficiency. Anaerobe 2017, 45, 50–58. [Google Scholar] [CrossRef]
- Calalang, J.; Cheung, H.; Lichimo, K.; So, B. Identifying Breed, Dietary, and Reproductive Factors Affecting the Gut Microbiome of Dogs with Inflammatory Bowel Disease. Undergrad. J. Exp. Microbiol. Immunol. 2021, 26, 1–13. [Google Scholar]
- Kell, D.B.; Kaprelyants, A.S.; Weichart, D.H.; Harwood, C.R.; Barer, M.R. Viability and Activity in Readily Culturable Bacteria: A Review and Discussion of the Practical Issues. Antonie Van Leeuwenhoek 1998, 73, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Pretorius, E. On the Translocation of Bacteria and Their Lipopolysaccharides between Blood and Peripheral Locations in Chronic, Inflammatory Diseases: The Central Roles of LPS and LPS-Induced Cell Death. Integr. Biol. 2015, 7, 1339–1377. [Google Scholar] [CrossRef] [Green Version]
- Innocente, G.; Patuzzi, I.; Furlanello, T.; Di Camillo, B.; Bargelloni, L.; Giron, M.C.; Facchin, S.; Savarino, E.; Azzolin, M.; Simionati, B. Machine Learning and Canine Chronic Enteropathies: A New Approach to Investigate FMT Effects. Vet. Sci. 2022, 9, 502. [Google Scholar] [CrossRef]
- Albenberg, L.; Esipova, T.V.; Judge, C.P.; Bittinger, K.; Chen, J.; Laughlin, A.; Grunberg, S.; Baldassano, R.N.; Lewis, J.D.; Li, H.; et al. Correlation Between Intraluminal Oxygen Gradient and Radial Partitioning of Intestinal Microbiota. Gastroenterology 2014, 147, 1055–1063.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swidsinski, A.; Weber, J.; Loening-Baucke, V.; Hale, L.P.; Lochs, H. Spatial Organization and Composition of the Mucosal Flora in Patients with Inflammatory Bowel Disease. J. Clin. Microbiol. 2005, 43, 3380–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G.; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462.e14. [Google Scholar] [CrossRef]
- Krajina, B.A.; Tropini, C.; Zhu, A.; DiGiacomo, P.; Sonnenburg, J.L.; Heilshorn, S.C.; Spakowitz, A.J. Dynamic Light Scattering Microrheology Reveals Multiscale Viscoelasticity of Polymer Gels and Precious Biological Materials. ACS Cent. Sci. 2017, 3, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Janney, A.; Powrie, F.; Mann, E.H. Host–Microbiota Maladaptation in Colorectal Cancer. Nature 2020, 585, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Potgieter, M.; Bester, J.; Kell, D.B.; Pretorius, E. The Dormant Blood Microbiome in Chronic, Inflammatory Diseases. FEMS Microbiol. Rev. 2015, 39, 567–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy | Sick | p-Value | ||||
|---|---|---|---|---|---|---|
| Median | SE | Median | SE | |||
| Actinobacteriota | Feces | 46.1 | 8.1 | 3.4 | 4.6 | 0.001 |
| Blood | 14.3 | 8.1 | 9.4 | 7.9 | 0.579 | |
| Bacteroidota | Feces | 38.1 | 13.6 | 6.4 | 2.6 | 0.005 |
| Blood | 2.4 | 16.2 | 167.2 | 7.0 | <0.0001 | |
| Firmicutes | Feces | 749.0 | 31.1 | 705.7 | 48.6 | 0.788 |
| Blood | 33.5 | 32.8 | 382.1 | 8.5 | <0.0001 | |
| Fusobacteriota | Feces | 42.0 | 10.8 | 4.8 | 5.1 | 0.038 |
| Blood | 0.0 | 0.2 | 2.9 | 0.7 | 0.000 | |
| Proteobacteria | Feces | 4.7 | 9.0 | 1.1 | 4.5 | 0.145 |
| Blood | 23.6 | 28.3 | 197.4 | 3.8 | <0.0001 | |
| Cyanobacteria | Feces | n.a. | n.a. | n.a. | n.a. | |
| Blood | 0.0 | 0.5 | 0.7 | 0.2 | 0.103 | |
| Healthy (Median) | SE | Sick (Median) | SE | H | p-Value | q-Value | Pseudo-F | |
|---|---|---|---|---|---|---|---|---|
| Shannon index | 7.60 | 0.52 | 8.56 | 0.14 | 4.78 | 0.029 | 0.029 | |
| Evenness index | 0.87 | 0.03 | 0.90 | 0.004 | 10.99 | 0.0009 | 0.0009 | |
| PERMANOVA weighted UniFrac | 0.001 | 0.001 | 30.56 | |||||
| PERMANOVA unweighted UniFrac | 0.001 | 0.001 | 4.93 |
| Healthy | Sick | p-Value | |||
|---|---|---|---|---|---|
| Median | SE | Median | SE | ||
| Acinetobacter | 7.7 | 4.1 | 0.0 | 0.1 | <0.0001 |
| Alistipes | 0.0 | 0.4 | 23.9 | 1.8 | <0.0001 |
| Alloprevotella | 0.0 | 0.4 | 9.8 | 1.0 | <0.0001 |
| Bacteroidales_RF16_group | 0.0 | 0.0 | 11.9 | 0.7 | <0.0001 |
| Bacteroides | 0.0 | 16.1 | 26.8 | 1.0 | 0.001 |
| Bifidobacterium | 0.3 | 0.6 | 3.1 | 7.0 | <0.0001 |
| Blautia | 0.0 | 1.8 | 4.6 | 1.8 | 0.010 |
| Christensenellaceae_R-7_group | 0.0 | 0.5 | 8.1 | 0.9 | <0.0001 |
| Clostridia_UCG-014 | 0.0 | 0.4 | 12.7 | 1.1 | <0.0001 |
| Collinsella | 0.0 | 7.1 | 6.5 | 1.8 | 0.011 |
| Cutibacterium | 6.2 | 5.7 | 0.0 | 0.2 | 0.000 |
| Escherichia-Shigella | 0.0 | 4.6 | 2.4 | 0.6 | 0.000 |
| Family_XIII_AD3011_group | 0.0 | 0.1 | 3.2 | 0.5 | <0.0001 |
| Fusobacterium | 0.0 | 0.2 | 2.9 | 0.7 | 0.000 |
| Izemoplasmatales | 0.0 | 0.1 | 2.7 | 0.4 | <0.0001 |
| Megamonas | 0.0 | 0.1 | 3.1 | 0.3 | <0.0001 |
| Monoglobus | 0.0 | 0.0 | 8.7 | 0.6 | <0.0001 |
| Muribaculaceae | 0.0 | 0.1 | 4.9 | 0.5 | <0.0001 |
| Prevotellaceae_UCG-001 | 0.0 | 0.0 | 2.2 | 0.3 | <0.0001 |
| Prevotellaceae_UCG-003 | 0.0 | 0.0 | 14.2 | 1.4 | <0.0001 |
| Rikenellaceae_RC9_gut_group | 0.0 | 0.4 | 35.7 | 2.3 | <0.0001 |
| Ruminobacter | 0.0 | 0.0 | 6.7 | 0.5 | <0.0001 |
| Ruminococcus | 0.0 | 0.1 | 6.8 | 0.6 | <0.0001 |
| Sphingomonas | 0.0 | 1.3 | 146.4 | 3.0 | <0.0001 |
| Turicibacter | 0.0 | 0.9 | 1.4 | 0.2 | 0.014 |
| UCG-005 | 0.0 | 0.5 | 56.5 | 3.1 | <0.0001 |
| UCG-010 | 0.0 | 0.0 | 84.6 | 6.1 | <0.0001 |
| [Eubacterium]_coprostanoligenes_group | 0.0 | 0.1 | 21.5 | 1.4 | <0.0001 |
| p-2534-18B5_gut_group | 0.0 | 0.1 | 2.3 | 0.3 | <0.0001 |
| Healthy (Median) | SE | Sick (Median) | SE | H | p-Value | q-Value | Pseudo-F | |
|---|---|---|---|---|---|---|---|---|
| Shannon index | 4.93 | 0.18 | 4.96 | 0.18 | 0.064 | 0.800 | 0.800 | |
| Evenness index | 0.66 | 0.02 | 0.70 | 0.02 | 1.099 | 0.294 | 0.294 | |
| PERMANOVA weighted UniFrac | 0.026 | 0.026 | 2.43 | |||||
| PERMANOVA unweighted UniFrac | 0.001 | 0.001 | 2.63 |
| Healthy | Sick | p-Value | |||
|---|---|---|---|---|---|
| Median | SE | Median | SE | ||
| Bacteroides | 17.9 | 9.9 | 2.9 | 1.2 | 0.001 |
| Collinsella | 33.4 | 8.4 | 0.0 | 4.0 | 0.000 |
| Fusobacterium | 42.0 | 10.8 | 4.8 | 5.1 | 0.038 |
| Megamonas | 5.0 | 3.5 | 0.1 | 34.3 | 0.002 |
| Slackia | 5.4 | 1.3 | 0.4 | 0.7 | 0.009 |
| Sutterella | 0.6 | 1.2 | 0.0 | 0.2 | 0.020 |
| [Ruminococcus]_gnavus_group | 16.0 | 7.0 | 4.9 | 3.0 | 0.011 |
| [Ruminococcus]_torques_group | 3.4 | 0.9 | 0.0 | 0.4 | 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarsella, E.; Meineri, G.; Sandri, M.; Ganz, H.H.; Stefanon, B. Characterization of the Blood Microbiome and Comparison with the Fecal Microbiome in Healthy Dogs and Dogs with Gastrointestinal Disease. Vet. Sci. 2023, 10, 277. https://doi.org/10.3390/vetsci10040277
Scarsella E, Meineri G, Sandri M, Ganz HH, Stefanon B. Characterization of the Blood Microbiome and Comparison with the Fecal Microbiome in Healthy Dogs and Dogs with Gastrointestinal Disease. Veterinary Sciences. 2023; 10(4):277. https://doi.org/10.3390/vetsci10040277
Chicago/Turabian StyleScarsella, Elisa, Giorgia Meineri, Misa Sandri, Holly H. Ganz, and Bruno Stefanon. 2023. "Characterization of the Blood Microbiome and Comparison with the Fecal Microbiome in Healthy Dogs and Dogs with Gastrointestinal Disease" Veterinary Sciences 10, no. 4: 277. https://doi.org/10.3390/vetsci10040277