
Next-Generation Sequencing for the Detection of Microbial Agents in Avian Clinical Samples


Abstract
:Simple Summary
Abstract
1. Introduction
2. Direct-Targeted and Non-Targeted NGS versus Classical Diagnostics
3. Short-Read Sequencing on Poultry and Avian Species
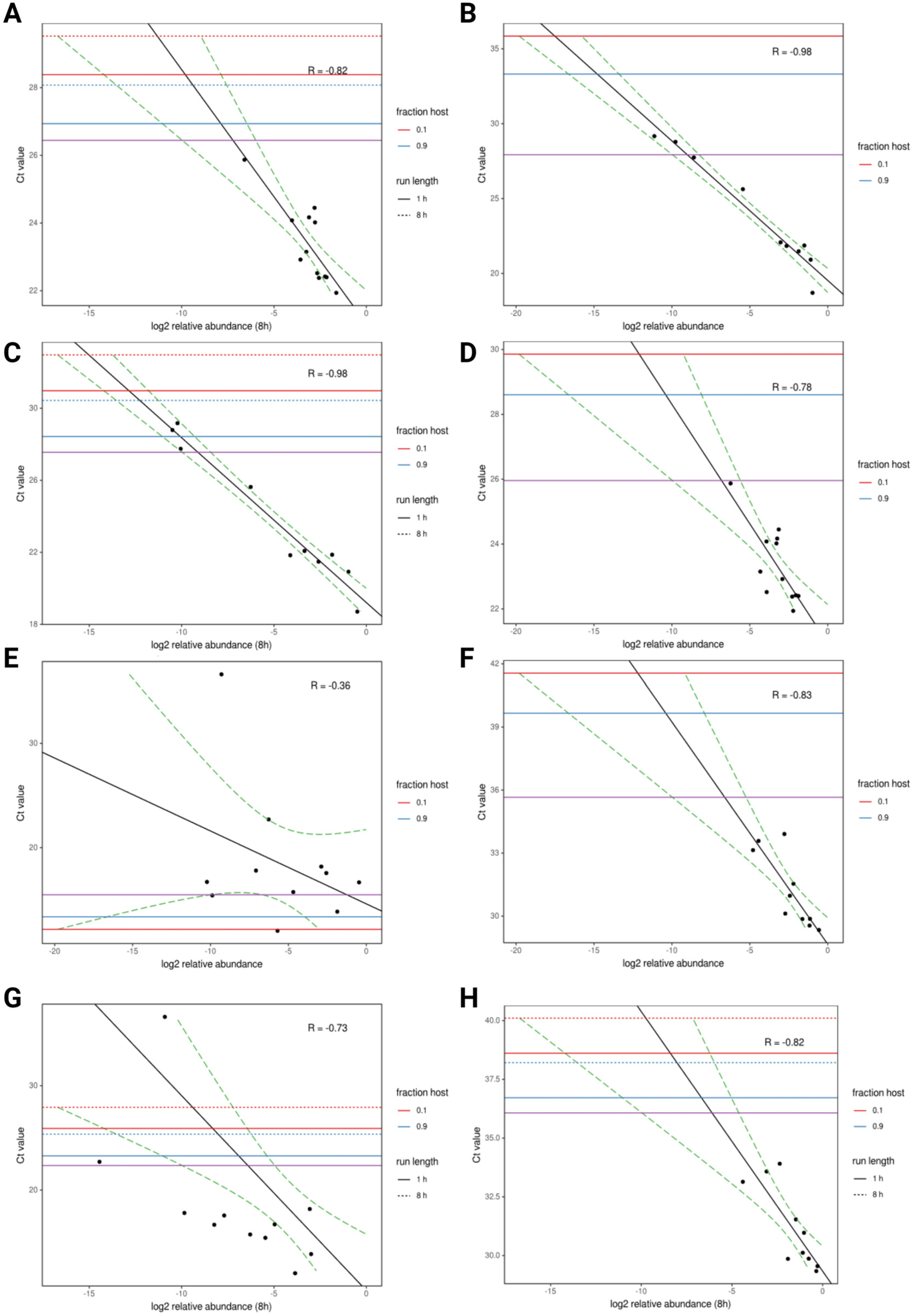
4. Long-Read Sequencing on Poultry and Avian Species
5. Direct Short and Long-Read Sequencing for Applications Not Related to Avian Disease Diagnostics
6. Challenges to the Adoption of NGS Diagnostics
7. Future Developments
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Borm, S.; Fu, Q.; Winand, R.; Vanneste, K.; Hakhverdyan, M.; Höper, D.; Vandenbussche, F. Evaluation of a Commercial Exogenous Internal Process Control for Diagnostic RNA Virus Metagenomics from Different Animal Clinical Samples. J. Virol. Methods 2020, 283, 113916. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Wang, J.; Granberg, F.; Colling, A. Next-Generation Sequencing Workflows in Veterinary Infection Biology: Towards Validation and Quality Assurance. Rev. Sci. Tech. 2016, 35, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Belák, S.; Freimanis, G.; Fusaro, A.; Granberg, F.; Höper, D.; King, D.P.; Monne, I.; Orton, R.; Rosseel, T. Next-Generation Sequencing in Veterinary Medicine: How Can the Massive Amount of Information Arising from High-Throughput Technologies Improve Diagnosis, Control, and Management of Infectious Diseases? Methods Mol. Biol. 2014, 1247, 415–436. [Google Scholar] [CrossRef]
- Howson, E.L.A.; Soldan, A.; Webster, K.; Beer, M.; Zientara, S.; Belák, S.; Sánchez-Vizcafno, J.M.; Van Borm, S.; King, D.P.; Fowler, V.L. Technological Advances in Veterinary Diagnostics: Opportunities to Deploy Rapid Decentralised Tests to Detect Pathogens Affecting Livestock. OIE Rev. Sci. Tech. 2017, 36, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Belák, S.; Karlsson, O.E.; Blomström, A.L.; Berg, M.; Granberg, F. New Viruses in Veterinary Medicine, Detected by Metagenomic Approaches. Vet. Microbiol. 2013, 165, 95–101. [Google Scholar] [CrossRef]
- Blomström, A.L. Viral Metagenomics as an Emerging and Powerful Tool in Veterinary Medicine. Vet. Q. 2011, 31, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Mathijs, E.; Vandenbussche, F.; Van Borm, S. Using Genomics for Surveillance of Veterinary Infectious Agents. OIE Rev. Sci. Tech. 2016, 35, 143–157. [Google Scholar] [CrossRef]
- Goodman, L.; Lahmers, K. Special Issue on Applied Next-Generation Sequencing in Veterinary Diagnostic Laboratories. J. Vet. Diagn. Investig. 2021, 33, 177–178. [Google Scholar] [CrossRef]
- Kumar, D. Next-Generation Sequencing as Diagnostic Tool in Veterinary Research. J. Anim. Res. 2019, 9, 797–806. [Google Scholar] [CrossRef]
- Belák, S.; Karlsson, O.E.; Leijon, M.; Granberg, F. High-Throughput Sequencing in Veterinary Infection Biology and Diagnostics. Rev. Sci. Tech. 2013, 32, 893–915. [Google Scholar] [CrossRef]
- Van Borm, S.; Obishakin, E.; Joannis, T.; Lambrecht, B.; van den Berg, T. Further Evidence for the Widespread Co-Circulation of Lineages 4b and 7 Velogenic Newcastle Disease Viruses in Rural Nigeria. Avian Pathol. 2012, 41, 377–382. [Google Scholar] [CrossRef]
- Dimitrov, K.M.; Sharma, P.; Volkening, J.D.; Goraichuk, I.V.; Wajid, A.; Rehmani, S.F.; Basharat, A.; Shittu, I.; Joannis, T.M.; Miller, P.J.; et al. A Robust and Cost-Effective Approach to Sequence and Analyze Complete Genomes of Small RNA Viruses. Virol. J. 2017, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Franca, M.; Howerth, E.W.; Carter, D.; Byas, A.; Poulson, R.; Afonso, C.L.; Stallknecht, D.E. Co-Infection of Mallards with Low-Virulence Newcastle Disease Virus and Low-Pathogenic Avian Influenza Virus. Avian Pathol. 2014, 43, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Lin, L.; Sebastian, A.; Lu, H. Detection and Characterization of Two Co-Infection Variant Strains of Avian Orthoreovirus (ARV) in Young Layer Chickens Using next-Generation Sequencing (NGS). Sci. Rep. 2016, 6, 24519. [Google Scholar] [CrossRef] [PubMed]
- Kariithi, H.M.; Volkening, J.D.; Leyson, C.M.; Afonso, C.L.; Christy, N.; Decanini, E.L.; Lemiere, S.; Suarez, D.L. Genome Sequence Variations of Infectious Bronchitis Virus Serotypes From Commercial Chickens in Mexico. Front. Vet. Sci. 2022, 9, 931272. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.G.; Patel, B.J.; Patel, S.S.; Raval, S.H.; Parmar, R.S.; Joshi, D.V.; Chauhan, H.C.; Chandel, B.S.; Patel, B.K. Metagenomic of Clinically Diseased and Healthy Broiler Affected with Respiratory Disease Complex. Data Brief. 2018, 19, 82–85. [Google Scholar] [CrossRef] [PubMed]
- van den Hoogen, B.G.; de Jong, J.C.; Groen, J.; Kuiken, T.; de Groot, R.; Fouchier, R.A.; Osterhaus, A.D. A Newly Discovered Human Pneumovirus Isolated from Young Children with Respiratory Tract Disease. Nat. Med. 2001, 7, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Kondov, N.O.; Deng, X.; Van Eenennaam, A.; Neibergs, H.L.; Delwart, E. A Metagenomics and Case-Control Study To Identify Viruses Associated with Bovine Respiratory Disease. J. Virol. 2015, 89, 5340–5349. [Google Scholar] [CrossRef] [PubMed]
- Rajeoni, A.H.; Ghalyanchilangeroudi, A.; Khalesi, B.; Madadi, M.S.; Hosseini, H. The Tracheal Virome of Broiler Chickens with Respiratory Disease Complex in Iran: The Metagenomics Study. Iran. J. Microbiol. 2021, 13, 337–344. [Google Scholar] [CrossRef]
- Diao, Z.; Han, D.; Zhang, R.; Li, J. Metagenomics Next-Generation Sequencing Tests Take the Stage in the Diagnosis of Lower Respiratory Tract Infections. J. Adv. Res. 2022, 38, 201–212. [Google Scholar] [CrossRef]
- Cibulski, S.; Alves de Lima, D.; Fernandes dos Santos, H.; Teixeira, T.F.; Tochetto, C.; Mayer, F.Q.; Roehe, P.M. A Plate of Viruses: Viral Metagenomics of Supermarket Chicken, Pork and Beef from Brazil. Virology 2021, 552, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, R.; Ravi, A.; Getino, M.; Pursley, I.; Horton, D.; Alikhan, N.-F.; Baker, D.; Gharbi, K.; Hall, N.; Watson, M.; et al. A Genomic Census of the Chicken Gut Microbiome Using Metagenomics and Culture. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Kwoka, K.T.T.; de Rooij, M.M.T.; Messink, A.B.; Wouters, I.M.; Smit, L.A.M.; Heederik, D.J.J.; Koopmans, M.P.G.; Phan, M.V.T. Comparative Viral Metagenomics from Chicken Feces and Farm Dust in the Netherlands. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gilroy, R.; Ravi, A.; Getino, M.; Pursley, I.; Horton, D.L.; Alikhan, N.F.; Baker, D.; Gharbi, K.; Hall, N.; Watson, M.; et al. Extensive Microbial Diversity within the Chicken Gut Microbiome Revealed by Metagenomics and Culture. PeerJ 2021, 9, e10941. [Google Scholar] [CrossRef]
- Kariithi, H.M.; Christy, N.; Decanini, E.L.; Lemiere, S.; Volkening, J.D.; Afonso, C.L.; Suarez, D.L. Detection and Genome Sequence Analysis of Avian Metapneumovirus Subtype A Viruses Circulating in Commercial Chicken Flocks in Mexico. Vet. Sci. 2022, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.L.; He, Y.; Zhang, J.; Dimitrov, K.M.; Sharma, P.; Miller, P.J.; Isidoro-Ayza, M.; Ip, H.S.; Fenton, H.; Poulson, R.L.; et al. Next-Generation Sequencing of Newcastle Disease Viruses from Formalin-Fixed Paraffin-Embedded Tissues. University of Georgia. 2019. Available online: http://getd.libs.uga.edu/pdfs/butt_salman-latif_201908_phd.pdf (accessed on 24 November 2023).
- Kokkat, T.J.; Patel, M.S.; McGarvey, D.; LiVolsi, V.A.; Baloch, Z.W. Archived Formalin-Fixed Paraffin-Embedded (FFPE) Blocks: A Valuable Underexploited Resource for Extraction of DNA, RNA, and Protein. Biopreserv. Biobank 2013, 11, 101–106. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Isidoro-Ayza, M.; Butt, S.L.; Sharma, P.; Dimitrov, K.M.; Afonso, C.L.; Ip, H.S.; Stanton, J.B. Detection and Sequencing of PPMV-1 in Paraffin-Embedded Tissues from Wild Pigeons by next-Generation Sequencing. In Proceedings of the American College of Veterinary Pathologists, Annual Meeting, New Orleans, LA, USA, 3–7 December 2016. [Google Scholar]
- Butt, S.L.; Dimitrov, K.M.; Zhang, J.; Wajid, A.; Bibi, T.; Basharat, A.; Brown, C.C.; Rehmani, S.F.; Stanton, J.B.; Afonso, C.L. Enhanced Phylogenetic Resolution of Newcastle Disease Outbreaks Using Complete Viral Genome Sequences from Formalin-Fixed Paraffin-Embedded Tissue Samples. Virus Genes. 2019, 55, 502–512. [Google Scholar] [CrossRef]
- De Oliveira, L.B.; Stanton, J.B.; Zhang, J.; Brown, C.; Butt, S.L.; Dimitrov, K.; Afonso, C.L.; Volkening, J.D.; Lara, L.J.C.; de Oliveira, C.S.F.; et al. Runting and Stunting Syndrome in Broiler Chickens: Histopathology and Association with a Novel Picornavirus. Vet. Pathol. 2021, 58, 123–135. [Google Scholar] [CrossRef]
- Blankenberg, D.; Kuster, G.V.; Coraor, N.; Ananda, G.; Lazarus, R.; Mangan, M.; Nekrutenko, A.; Taylor, J. Galaxy: A Web-Based Genome Analysis Tool for Experimentalists. Curr. Protoc. Mol. Biol. 2010, 89. [Google Scholar] [CrossRef]
- Jalili, V.; Afgan, E.; Gu, Q.; Clements, D.; Blankenberg, D.; Goecks, J.; Taylor, J.; Nekrutenko, A. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2020 Update. Nucleic Acids Res. 2021, 48, W395–W402. [Google Scholar] [CrossRef]
- Sharma, P.; Killmaster, L.F.; Volkening, J.D.; Cardenas-Garcia, S.; Shittu, I.; Meseko, C.A.; Sulaiman, L.K.; Joannis, T.M.; Miller, P.J.; Afonso, C.L. Draft Genome Sequences of Five Novel Ochrobactrum spp. Isolated from Different Avian Hosts in Nigeria. Genome Announc. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Steensels, M.; Mathijs, E.; Vandenbussche, F.; van den Berg, T.; Lambrecht, B. Metagenomic Sequencing Determines Complete Infectious Bronchitis Virus (Avian Gammacoronavirus) Vaccine Strain Genomes and Associated Viromes in Chicken Clinical Samples. Virus Genes. 2021, 57, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Kariithi, H.M.; Welch, C.N.; Ferreira, H.L.; Pusch, E.A.; Ateya, L.O.; Binepal, Y.S.; Apopo, A.A.; Dulu, T.D.; Afonso, C.L.; Suarez, D.L. Genetic Characterization and Pathogenesis of the First H9N2 Low Pathogenic Avian Influenza Viruses Isolated from Chickens in Kenyan Live Bird Markets. Infect. Genet. Evol. 2020, 78, 104074. [Google Scholar] [CrossRef] [PubMed]
- Kariithi, H.M.; Ferreira, H.L.; Welch, C.N.; Ateya, L.O.; Apopo, A.A.; Zoller, R.; Volkening, J.D.; Williams-Coplin, D.; Parris, D.J.; Olivier, T.L.; et al. Surveillance and Genetic Characterization of Virulent Newcastle Disease Virus Subgenotype V.3 in Indigenous Chickens from Backyard Poultry Farms and Live Bird Markets in Kenya. Viruses 2021, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Youk, S.; Lee, D.H.; Ferreira, H.L.; Afonso, C.L.; Absalon, A.E.; Swayne, D.E.; Suarez, D.L.; Pantin-Jackwood, M.J. Rapid Evolution of Mexican H7N3 Highly Pathogenic Avian Influenza Viruses in Poultry. PLoS ONE 2019, 14, e0222457. [Google Scholar] [CrossRef] [PubMed]
- Youk, S.; Leyson, C.M.; Parris, D.J.; Kariithi, H.M.; Suarez, D.L.; Pantin-Jackwood, M.J. Phylogenetic Analysis, Molecular Changes, and Adaptation to Chickens of Mexican Lineage H5N2 Low-Pathogenic Avian Influenza Viruses from 1994 to 2019. Transbound. Emerg. Dis. 2022, 69, E1445–E1459. [Google Scholar] [CrossRef]
- Sabra, M.; Dimitrov, K.M.; Goraichuk, I.V.; Wajid, A.; Sharma, P.; Williams-Coplin, D.; Basharat, A.; Rehmani, S.F.; Muzyka, D.V.; Miller, P.J.; et al. Phylogenetic Assessment Reveals Continuous Evolution and Circulation of Pigeon-Derived Virulent Avian Avulaviruses 1 in Eastern Europe, Asia, and Africa. BMC Vet. Res. 2017, 13, 291. [Google Scholar] [CrossRef]
- Goraichuk, I.V.; Kulkarni, A.B.; Williams-Coplin, D.; Suarez, D.L.; Afonso, C.L.; Roux, S. First Complete Genome Sequence of Currently Circulating Infectious Bronchitis Virus Strain DMV/1639 of the GI-17 Lineage. Microbiol. Resour. Announc. 2019, 8, e00840-19. [Google Scholar] [CrossRef]
- Goraichuk, I.V.; Williams-Coplin, D.; Wibowo, M.H.; Durr, P.A.; Asmara, W.; Artanto, S.; Dimitrov, K.M.; Afonso, C.L.; Suarez, D.L.; Roux, S. Complete Genome Sequences of 11 Newcastle Disease Virus Isolates of Subgenotype VII.2 from Indonesia. Microbiol. Resour. Announc. 2020, 9. [Google Scholar] [CrossRef]
- Goraichuk, I.V.; Davis, J.F.; Parris, D.J.; Kariithi, H.M.; Afonso, C.L.; Suarez, D.L. Near-Complete Genome Sequences of Five Siciniviruses from North America. Microbiol. Resour. Announc. 2021, 10. [Google Scholar] [CrossRef]
- Goraichuk, I.V.; Davis, J.F.; Kulkarni, A.B.; Afonso, C.L.; Suarez, D.L.; Roux, S. Complete Genome Sequence of Avian Coronavirus Strain GA08 (GI-27 Lineage). Microbiol. Resour. Announc. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Goraichuk, I.V.; Dimitrov, K.M.; Sharma, P.; Miller, P.J.; Swayne, D.E.; Suarez, D.L.; Afonso, C.L. Complete Genome Sequences of Four Avian Paramyxoviruses of Serotype 10 Isolated from Rockhopper Penguins on the Falkland Islands. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Goraichuk, I.V.; Davis, J.F.; Kulkarni, A.B.; Afonso, C.L.; Suarez, D.L. A 24-Year-Old Sample Contributes the Complete Genome Sequence of Fowl Aviadenovirus D from the United States. Microbiol. Resour. Announc. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Goraichuk, I.V.; Davis, J.F.; Afonso, C.L.; Suarez, D.L. Complete Coding Sequences of Three Chicken Parvovirus Isolates from the United States. Microbiol. Resour. Announc. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Shah-Majid, M. Effect of Mixed Infection of Mycoplasma Gallinarum and Newcastle Disease Virus (F Strain) on the Tracheal Epithelium of Village Chickens. Res. Vet. Sci. 1996, 61, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.; Awais, M.M.; Anwar, M.I.; Ehtisham-ul-Haque, S.; Nasir, A.; Saleemi, M.K.; Ashraf, K. The Effect of Infection with Mixed Eimeria Species on Hematology and Immune Responses Following Newcastle Disease and Infectious Bursal Disease Booster Vaccination in Broilers. Vet. Q. 2015, 35, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nonomura, I.; Shimizu, F.; Shoya, S.; Horiuchi, T. Mixed Infection with Mycoplasma Gallisepticum and the B1 Strain of Newcastle Disease Virus in Chickens. Natl. Inst. Anim. Health Q 1970, 10, 58–65. [Google Scholar]
- Cardenas-Garcia, S.; Sharma, P.; Shittu, I.; Joannis, T.M.; Volkening, J.D.; Williams-Coplin, D.; Miller, P.J.; Dimitrov, K.M.D.; Ficht, T.; Afonso, C.L. Identification of a Putative Novel Brucella species by Next-Generation Sequencing from Samples Collected in Nigeria from Different Avian Hosts. In The Middle East and South Asia Conference on Epigenetics and Genomics of Infectious Diseases. 2016. Available online: https://www.gryphonscientific.com/our-work/middle-east-and-south-asia-conference-on-epigenetics-and-genomics-of-infectious-diseases/ (accessed on 24 November 2023).
- Sharma, P.; Killmaster, L.F.; Volkening, J.D.; Cardenas-Garcia, S.; Wajid, A.; Rehmani, S.F.; Basharat, A.; Miller, P.J.; Afonso, C.L. Draft Genome Sequences of Three Ochrobactrum spp. Isolated from Different Avian Hosts in Pakistan. Genome Announc. 2018, 6. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Goecks, J.; Nekrutenko, A.; Taylor, J. Galaxy: A Comprehensive Approach for Supporting Accessible, Reproducible, and Transparent Computational Research in the Life Sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef]
- Kariithi, H.M.; Volkening, J.D.; Alves, V.V.; Reis-Cunha, J.L.; Arantes, L.C.R.V.; Fernando, F.S.; Filho, T.F.; da Silva Martins, N.R.; Lemiere, S.; de Freitas Neto, O.C.; et al. Complete Genome Sequences of Avian Metapneumovirus Subtype B Vaccine Strains from Brazil. Microbiol. Resour. Announc. 2023, 12, e0023523. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.T.V.; Irinyi, L.; Hu, Y.; Schwessinger, B.; Meyer, W. Long-Reads-Based Metagenomics in Clinical Diagnosis With a Special Focus on Fungal Infections. Front. Microbiol. 2022, 12, 708550. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.L.; Erwood, E.C.; Zhang, J.; Sellers, H.S.; Young, K.; Lahmers, K.K.; Stanton, J.B. Real-Time, MinION-Based, Amplicon Sequencing for Lineage Typing of Infectious Bronchitis Virus from Upper Respiratory Samples. J. Vet. Diagn. Investig. 2021, 33, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Spatz, S.J.; Garcia, M.; Riblet, S.; Ross, T.A.; Volkening, J.D.; Taylor, T.L.; Kim, T.; Afonso, C.L. MinION Sequencing to Genotype US Strains of Infectious Laryngotracheitis Virus. Avian Pathol. 2019, 48, 255–269. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Harder, T.; Beer, M.; Pohlmann, A. Rapid Multiplex MinION Nanopore Sequencing Workflow for Influenza A Viruses. BMC Infect. Dis. 2020, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, H.; Mao, L.; Yu, H.; Yu, X.; Sun, Z.; Qian, X.; Cheng, S.; Chen, S.; Chen, J.; et al. Rapid Genomic Characterization of SARS-CoV-2 Viruses from Clinical Specimens Using Nanopore Sequencing. Sci. Rep. 2020, 10, 17492. [Google Scholar] [CrossRef] [PubMed]
- Arana, C.; Liang, C.; Brock, M.; Zhang, B.; Zhou, J.; Chen, L.; Cantarel, B.; SoRelle, J.; Hooper, L.V.; Raj, P. A Short plus Long-Amplicon Based Sequencing Approach Improves Genomic Coverage and Variant Detection in the SARS-CoV-2 Genome. PLoS ONE 2022, 17, e0261014. [Google Scholar] [CrossRef]
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M.; et al. Evaluation of NGS-Based Approaches for SARS-CoV-2 Whole Genome Characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef]
- Yip, C.C.-Y.; Chan, W.-M.; Ip, J.D.; Seng, C.W.-M.; Leung, K.-H.; Poon, R.W.-S.; Ng, A.C.-K.; Wu, W.-L.; Zhao, H.; Chan, K.-H. Nanopore Sequencing Reveals Novel Targets for Detection and Surveillance of Human and Avian Influenza A Viruses. J. Clin. Microbiol. 2020, 58. [Google Scholar] [CrossRef]
- Crossley, B.M.; Rejmanek, D.; Baroch, J.; Stanton, J.B.; Young, K.T.; Killian, M.L.; Torchetti, M.K.; Hietala, S.K. Nanopore Sequencing as a Rapid Tool for Identification and Pathotyping of Avian Influenza A viruses. J. Veter-Diagn. Investig. 2021, 33, 253–260. [Google Scholar] [CrossRef]
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J. Metagenomic Nanopore Sequencing of Influenza Virus Direct from Clinical Respiratory Samples. J. Clin. Microbiol. 2019, 58, e00963-19. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.L.; Kariithi, H.M.; Volkening, J.D.; Taylor, T.L.; Leyson, C.; Pantin-Jackwood, M.; Suarez, D.L.; Stanton, J.B.; Afonso, C.L. Comparable Outcomes from Long and Short Read Random Sequencing of Total RNA for Detection of Pathogens in Chicken Respiratory Samples. Front. Veter-Sci. 2022, 9, 1073919. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.L.; Taylor, T.L.; Volkening, J.D.; Dimitrov, K.M.; Williams-Coplin, D.; Lahmers, K.K.; Miller, P.J.; Rana, A.M.; Suarez, D.L.; Afonso, C.L.; et al. Rapid Virulence Prediction and Identification of Newcastle Disease Virus Genotypes Using Third-Generation Sequencing. Virol. J. 2018, 15, 179. [Google Scholar] [CrossRef] [PubMed]
- Kogut, M.H. Role of Diet-Microbiota Interactions in Precision Nutrition of the Chicken: Facts, Gaps, and New Concepts. Poult. Sci. 2022, 101, 101673. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Lee, T.K.; Sul, W.J. Metagenomic Analysis of Chicken Gut Microbiota for Improving Metabolism and Health of Chickens—A Review. Asian-Australas. J. Anim. Sci. 2015, 28, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Delwart, E. What is for Dinner? Viral Metagenomics of US Store Bought Beef, Pork, and Chicken. Virology 2014, 468, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Kurian, S.M.; Gordon, S.; Barrick, B.; Dadlani, M.N.; Fanelli, B.; Cornell, J.B.; Head, S.R.; Marsh, C.L.; Case, J. Feasibility and Comparison Study of Fecal Sample Collection Methods in Healthy Volunteers and Solid Organ Transplant Recipients Using 16S rRNA and Metagenomics Approaches. Biopreserv. Biobanking 2020, 18, 425–440. [Google Scholar] [CrossRef]
- Hemamalini, N.; Shanmugam, S.A.; Kathirvelpandian, A.; Deepak, A.; Kaliyamurthi, V.; Suresh, E. A Critical Review on the Antimicrobial Resistance, Antibiotic Residue and Metagenomics-Assisted Antimicrobial Re-sistance Gene Detection in Freshwater Aquaculture Environment. Aquac. Res. 2022, 53, 344–366. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R.; Kalaba, M.; Kosutic, J.; Godman, B.; Radonjic, V.; Vujic, A.; Jankovic, S.; Srebro, D.; Kalaba, Z.; et al. Insights into Antibiotic Resistance through Metagenomic Approaches. Futur. Microbiol. 2012, 7, 73–89. [Google Scholar] [CrossRef]
- De, R. Metagenomics: Aid to Combat Antimicrobial Resistance in Diarrhea. Gut Pathog. 2019, 11, 47. [Google Scholar] [CrossRef]
- Jing, R.; Yan, Y. Metagenomic Analysis Reveals Antibiotic Resistance Genes in the Bovine Rumen. Microb. Pathog. 2020, 149, 104350. [Google Scholar] [CrossRef] [PubMed]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A Deep Learning Approach for Predicting Antibiotic Resistance Genes from Metagenomic Data. Microbiome 2018, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Skarżyńska, M.; Leekitcharoenphon, P.; Hendriksen, R.S.; Aarestrup, F.M.; Wasyl, D. A metagenomic Glimpse into the Gut of Wild and Domestic Animals: Quantification of Antimicrobial Resistance and More. PLoS ONE 2020, 15, e0242987. [Google Scholar] [CrossRef] [PubMed]
- Namkung, J. Machine Learning Methods for Microbiome Studies. J. Microbiol. 2020, 58, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Soueidan, H.; Nikolski, M. Machine Learning for Metagenomics: Methods and Tools. Metagenomics 2016, 1, 1–19. [Google Scholar] [CrossRef]
- Cheung, H.; Yu, J. Machine Learning on Microbiome Research in Gastrointestinal Cancer. J. Gastroenterol. Hepatol. 2021, 36, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Krause, T.; Wassan, J.T.; Mc Kevitt, P.; Wang, H.; Zheng, H.; Hemmje, M. Analyzing Large Microbiome Datasets Using Machine Learning and Big Data. BioMedInformatics 2021, 1, 138–165. [Google Scholar] [CrossRef]
- Manandhar, I.; Alimadadi, A.; Aryal, S.; Munroe, P.B.; Joe, B.; Cheng, X. Gut Microbiome-Based Supervised Machine Learning for Clinical Diagnosis of Inflammatory Bowel Diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G328–G337. [Google Scholar] [CrossRef]
- Curry, K.D.; Nute, M.G.; Treangen, T.J. It Takes Guts to Learn: Machine Learning Techniques for Disease Detection from the Gut Microbiome. Emerg. Top. Life Sci. 2021, 5, 815–827. [Google Scholar] [CrossRef]
- Bolinger, H.; Tran, D.; Harary, K.; Paoli, G.C.; Guron, G.K.P.; Namazi, H.; Khaksar, R. Utilizing the Microbiota and Machine Learning Algorithms To Assess Risk of Salmonella Contamination in Poultry Rinsate. J. Food Prot. 2021, 84, 1648–1657. [Google Scholar] [CrossRef]
- Thibodeau, A.; Fravalo, P.; Yergeau, E.; Arsenault, J.; Lahaye, L.; Letellier, A. Chicken Caecal Microbiome Modifications Induced by Campylobacter jejuni Colonization and by a Non-Antibiotic Feed Additive. PLoS ONE 2015, 10, e0131978. [Google Scholar] [CrossRef] [PubMed]
- Awad, W.A.; Mann, E.; Dzieciol, M.; Hess, C.; Schmitz-Esser, S.; Wagner, M.; Hess, M. Age-Related Differences in the Luminal and Mucosa-Associated Gut Microbiome of Broiler Chickens and Shifts Associated with Campylobacter jejuni Infection. Front. Cell. Infect. Microbiol. 2016, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Walugembe, M.; Hsieh, J.C.F.; Koszewski, N.J.; Lamont, S.J.; Persia, M.E.; Rothschild, M.F. Effects of Dietary Fiber on Cecal Short-Chain Fatty Acid and Cecal Microbiota of Broiler and Laying-Hen Chicks. Poult. Sci. 2015, 94, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.L.; Polson, S.W.; Lee, K.H. K-Mer-Based Metagenomics Tools Provide a Fast and Sensitive Approach for the Detection of Viral Contaminants in Biopharmaceutical and Vaccine Manufacturing Applications Using Next-Generation Sequencing. mSphere 2021, 6, 110–128. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Benetti, L.; Blumel, J.; Deforce, D.; Egan, W.M.; Knezevic, I.; Krause, P.R.; Mallet, L.; Mayer, D.; Minor, P.D.; et al. Report of the International Conference on next Generation Sequencing for Adventitious Virus Detection in Biologicals. Biologicals 2018, 55, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Blümel, J.; Deforce, D.; Gruber, M.F.; Jungbäck, C.; Knezevic, I.; Mallet, L.; Mackay, D.; Matthijnssens, J.; O’Leary, M.; et al. Report of the Second International Conference on next Generation Sequencing for Adventitious Virus Detection in Biologics for Humans and Animals. Biologicals 2020, 67, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Dourou, D.; Spyrelli, E.D.; Doulgeraki, A.I.; Argyri, A.A.; Grounta, A.; Nychas, G.-J.E.; Chorianopoulos, N.G.; Tassou, C.C. Microbiota of Chicken Breast and Thigh Fillets Stored under Different Refrigeration Temperatures Assessed by Next-Generation Sequencing. Foods 2021, 10, 765. [Google Scholar] [CrossRef]
- Li, S.; Mann, D.A.; Zhang, S.; Qi, Y.; Meinersmann, R.J.; Deng, X. Microbiome-Informed Food Safety and Quality: Longitudinal Consistency and Cross-Sectional Distinctiveness of Retail Chicken Breast Microbiomes. mSystems 2020, 5, e00589-20. [Google Scholar] [CrossRef]
- Billington, C.; Kingsbury, J.M.; Rivas, L. Metagenomics Approaches for Improving Food Safety: A Review. J. Food Prot. 2022, 85, 448–464. [Google Scholar] [CrossRef]
- Sabater, C.; Cobo-Díaz, J.F.; Álvarez-Ordóñez, A.; Ruas-Madiedo, P.; Ruiz, L.; Margolles, A. Novel Methods of Microbiome Analysis in the Food Industry. Int. Microbiol. 2021, 24, 593–605. [Google Scholar] [CrossRef]
- Jagadeesan, B.; Gerner-Smidt, P.; Allard, M.W.; Leuillet, S.; Winkler, A.; Xiao, Y.; Chaffron, S.; Van Der Vossen, J.; Tang, S.; Katase, M.; et al. The Use of Next Generation Sequencing for Improving Food Safety: Translation into Practice. Food Microbiol. 2019, 79, 96–115. [Google Scholar] [CrossRef]
- Josefsen, M.H.; Andersen, S.C.; Christensen, J.; Hoorfar, J. Microbial Food Safety: Potential of DNA Extraction Methods for Use in Diagnostic Metagenomics. J. Microbiol. Methods 2015, 114, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Szarvas, J.; Ahrenfeldt, J.; Cisneros, J.L.B.; Thomsen, M.C.F.; Aarestrup, F.M.; Lund, O. Large Scale Automated Phylogenomic Analysis of Bacterial Isolates and the Evergreen Online Platform. Commun. Biol. 2020, 3, 137. [Google Scholar] [CrossRef] [PubMed]
- Saenz-García, C.E.; Castañeda-Serrano, P.; Silva, E.M.M.; Alvarado, C.Z.; Nava, G.M. Insights into the Identification of the Specific Spoilage Organisms in Chicken Meat. Foods 2020, 9, 225. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.; Li, J.; Zhang, Y.; Pu, H.; Yan, X. Application of Next Generation Sequencing-Based Rapid Detection Platform for Microbiological Diagnosis and Drug Resistance Prediction in Acute Lower Respiratory Infection. Ann. Transl. Med. 2020, 8, 1644. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, L.; Jiang, H. Integrated Bioinformatics and Clinical Correlation Analysis of Key Genes, Pathways, and Potential Therapeutic Agents Related to Diabetic Nephropathy. Dis. Markers 2022, 2022, 9204201. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, C.; Wu, Q.; Wang, J.; Tang, X.; Wu, Z.; Tang, L.; Zhou, Y. Systematical Analysis of Underlying Markers Associated with Marfan Syndrome via Integrated Bioinformatics and Machine Learning Strategies. J. Biomol. Struct. Dyn. 2023, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.L.; Volkening, J.D.; DeJesus, E.; Simmons, M.; Dimitrov, K.M.; Tillman, G.E.; Suarez, D.L.; Afonso, C.L. Rapid, Multiplexed, Whole Genome and Plasmid Sequencing of Foodborne Pathogens Using Long-Read Nanopore Technology. Sci. Rep. 2019, 9, 16350. [Google Scholar] [CrossRef]
- Forbes, J.D.; Knox, N.C.; Ronholm, J.; Pagotto, F.; Reimer, A. Metagenomics: The next Culture-Independent Game Changer. Front. Microbiol. 2017, 8, 1069. [Google Scholar] [CrossRef]
- Bikel, S.; Valdez-Lara, A.; Cornejo-Granados, F.; Rico, K.; Canizales-Quinteros, S.; Soberón, X.; Del Pozo-Yauner, L.; Ochoa-Leyva, A. Combining metagenomics, metatranscriptomics and viromics to explore novel micro-bial interactions: Towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 2015, 13, 390–401. [Google Scholar] [CrossRef]
- Hasan, M.R.; Sundararaju, S.; Tang, P.; Tsui, K.-M.; Lopez, A.P.; Janahi, M.; Tan, R.; Tilley, P. A Metagenomics-Based Diagnostic Approach for Central Nervous System Infections in Hospital Acute Care Setting. Sci. Rep. 2020, 10, 11194. [Google Scholar] [CrossRef] [PubMed]
- Tourlousse, D.M.; Narita, K.; Miura, T.; Sakamoto, M.; Ohashi, A.; Shiina, K.; Matsuda, M.; Miura, D.; Shimamura, M.; Ohyama, Y.; et al. Validation and Standardization of DNA Extraction and Library Construction Methods for Metagenomics-Based Human Fecal Microbiome Measurements. Microbiome 2021, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis. Evol. Bioinform. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Afshinnekoo, E.; Chou, C.; Alexander, N.; Ahsanuddin, S.; Schuetz, A.N.; Mason, C.E. Precision Metagenomics: Rapid Metagenomic Analyses for Infectious Disease Diagnostics and Public Health Surveillance. J. Biomol. Tech. JBT 2017, 28, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Nazir, A. Review on Metagenomics and Its Applications. Imp. J. Interdiscip. Res. 2016, 2, 277–286. [Google Scholar]
- Zhang, Y.-Z.; Chen, Y.-M.; Wang, W.; Qin, X.-C.; Holmes, E.C. Expanding the RNA Virosphere by Unbiased Metagenomics. Annu. Rev. Virol. 2019, 6, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Schlaberg, R. Metagenomics for the Diagnosis of Exotic Infections. Int. J. Infect. Dis. 2018, 73, 58. [Google Scholar] [CrossRef]
- Graf, E.H.; Simmon, K.E.; Tardif, K.D.; Hymas, W.; Flygare, S.; Eilbeck, K.; Yandell, M.; Schlaberg, R. Unbiased Detection of Respiratory Viruses by Use of RNA Sequencing-Based Metagenomics: A Systematic Comparison to a Commercial PCR Panel. J. Clin. Microbiol. 2016, 54, 1000–1007. [Google Scholar] [CrossRef]
- Kwok, K.T.T.; Nieuwenhuijse, D.F.; Phan, M.V.T.; Koopmans, M.P.G. Virus Metagenomics in Farm Animals: A Systematic Review. Viruses 2020, 12, 107. [Google Scholar] [CrossRef]
- Mitchell, S.L. Use of Diagnostic Metagenomics in the Clinical Microbiology Laboratory. Am. Soc. Clin. Lab. Sci. 2019, 32. [Google Scholar] [CrossRef]
- Peterson, C.-L.; Alexander, D.; Chen, J.C.-Y.; Adam, H.; Walker, M.; Ali, J.; Forbes, J.; Taboada, E.; Barker, D.O.R.; Graham, M.; et al. Clinical Metagenomics Is Increasingly Accurate and Affordable to Detect Enteric Bacterial Pathogens in Stool. Microorganisms 2022, 10, 441. [Google Scholar] [CrossRef] [PubMed]
- Olson, N.D.; Treangen, T.J.; Hill, C.M.; Cepeda-Espinoza, V.; Ghurye, J.; Koren, S.; Pop, M. Metagenomic Assembly through the Lens of Validation: Recent Advances in Assessing and Improving the Quality of Genomes Assembled from Metagenomes. Brief. Bioinform. 2018, 20, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Schlaberg, R.; Chiu, C.Y.; Miller, S.; Procop, G.W.; Weinstock, G. Validation of Metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection. Arch. Pathol. Lab. Med. 2017, 141, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, D.W.; Zagordi, O.; Geissberger, F.-D.; Kufner, V.; Schmutz, S.; Böni, J.; Metzner, K.J.; Trkola, A.; Huber, M. Optimization and Validation of Sample Preparation for Metagenomic Sequencing of Viruses in Clinical Samples. Microbiome 2017, 5, 94. [Google Scholar] [CrossRef]
- Blauwkamp, T.A.; Thair, S.; Rosen, M.J.; Blair, L.; Lindner, M.S.; Vilfan, I.D.; Kawli, T.; Christians, F.C.; Venkatasubrahmanyam, S.; Wall, G.D.; et al. Analytical and Clinical Validation of a Microbial Cell-Free DNA Sequencing Test for Infectious Disease. Nat. Microbiol. 2019, 4, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, Q.; Sheng, Y.; Wang, Q.; Wu, J.; Qiu, Z.; Lan, R.; Wang, Y.; Liu, Y. Metagenomics Combined with Comprehensive Validation as a Public Health Risk Assessment Tool for Urban and Agricultural Run-Off. Water Res. 2022, 209, 117941. [Google Scholar] [CrossRef] [PubMed]
- Halpin, K.; Tribolet, L.; Hobbs, E.C.; Singanallur, N.B. Perspectives and Challenges in Validating New Diagnostic Technologies. OIE Rev. Sci. Tech. 2021, 40, 145–157. [Google Scholar] [CrossRef]
- Rosseel, T.; Ozhelvaci, O.; Freimanis, G.; Van Borm, S. Evaluation of Convenient Pretreatment Protocols for RNA Virus Metagenomics in Serum and Tissue Samples. J. Virol. Methods 2015, 222, 72–80. [Google Scholar] [CrossRef]
- Rosseel, T.; Pardon, B.; De Clercq, K.; Ozhelvaci, O.; Van Borm, S. False-Positive Results in Metagenomic Virus Discovery: A Strong Case for Follow-Up Diagnosis. Transbound. Emerg. Dis. 2014, 61, 293–299. [Google Scholar] [CrossRef]
- Parris, D.J.; Kariithi, H.; Suarez, D.L. Non-Target RNA Depletion Strategy to Improve Sensitivity of Next-Generation Sequencing for the Detection of RNA Viruses in Poultry. J. Veter-Diagn. Investig. 2022, 34, 638–645. [Google Scholar] [CrossRef]
- Ganda, E.; Beck, K.L.; Haiminen, N.; Silverman, J.D.; Kawas, B.; Cronk, B.D.; Anderson, R.R.; Goodman, L.B.; Wiedmann, M. DNA Extraction and Host Depletion Methods Significantly Impact and Potentially Bias Bacterial Detection in a Biological Fluid. mSystems 2021, 6, e0061921. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; He, X.; Hoadley, K.A.; Parker, J.S.; Hayes, D.N.; Perou, C.M. Comparison of RNA-Seq by Poly (A) Capture, Ribosomal RNA Depletion, and DNA Microarray for Expression Profiling. BMC Genom. 2014, 15, 419. [Google Scholar] [CrossRef] [PubMed]
- Kafetzopoulou, L. Metagenomic Next Generation Sequencing for Viral Pathogens: Application and Validation of a Deployable Sequencer for Virus Identification. Ph.D. Thesis, University of Liverpool, Liverpool, UK, 2020. [Google Scholar]
- Crone, M.A.; Priestman, M.; Ciechonska, M.; Jensen, K.; Sharp, D.J.; Anand, A.; Randell, P.; Storch, M.; Freemont, P.S. A Role for Biofoundries in Rapid Development and Validation of Automated SARS-CoV-2 Clinical Diagnostics. Nat. Commun. 2020, 11, 4464. [Google Scholar] [CrossRef] [PubMed]
- van Boheemen, S.; van Rijn, A.L.; Pappas, N.; Carbo, E.C.; Vorderman, R.H.P.; Sidorov, I.; van’t Hof, P.J.; Mei, H.; Claas, E.C.J.; Kroes, A.C.M.; et al. Retrospective Validation of a Metagenomic Sequencing Protocol for Combined Detection of RNA and DNA Viruses Using Respiratory Samples from Pediatric Patients. J. Mol. Diagn. 2020, 22, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Espindola, A.S.; Cardwell, K.; Martin, F.N.; Hoyt, P.R.; Marek, S.M.; Schneider, W.; Garzon, C.D. A Step Towards Validation of High-Throughput Sequencing for the Identification of Plant Pathogenic Oomycetes. Phytopathology 2022, 112, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory Validation of a Clinical Metagenomic Sequencing Assay for Pathogen Detection in Cerebrospinal Fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.B.; Engberg, R.M.; Skov, J. A New Tool for Air Sample-Based Surveillance of Campylobacter and Salmonella in Poultry Flocks. J. Appl. Poult. Res. 2022, 31, 100236. [Google Scholar] [CrossRef]
- Hietala, S.K.; Hullinger, P.J.; Crossley, B.M.; Kinde, H.; Ardans, A.A. Environmental Air Sampling to Detect Exotic Newcastle Disease Virus in Two California Commercial Poultry Flocks. J. Veter-Diagn. Investig. 2005, 17, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Broza, Y.Y.; Haick, H. Biodiagnostics in an Era of Global Pandemics—From Biosensing Materials to Data Management. View 2022, 3, 20200164. [Google Scholar] [CrossRef]
- Lai, K.; Twine, N.; O’Brien, A.; Guo, Y.; Bauer, D. Artificial Intelligence and Machine Learning in Bioinformatics. In Encyclopedia of Bioinformatics and Computational Biology: ABC of Bioinformatics; Elsevier: Amsterdam, The Netherlands, 2018; Volume 1–3. [Google Scholar]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef]
- Mathieu, A.; Leclercq, M.; Sanabria, M.; Perin, O.; Droit, A. Machine Learning and Deep Learning Applications in Metagenomic Taxonomy and Functional Annotation. Front. Microbiol. 2022, 13, 811495. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, G.; Yu, J.; Sung, J.J.Y. Artificial Intelligence and Metagenomics in Intestinal Diseases. J. Gastroenterol. Hepatol. 2021, 36, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Baugher, D.; Larsen, A.W.; Chen, Y.; Icenhour, C.R.; Valencia, C.A. Female Urinary Microbiome Analysis and Artificial Intelligence Enhances the Infectious Diagnostic Yield in Precision Medicine. Microbiol. Infect. Dis. 2021, 5, 1–8. [Google Scholar] [CrossRef]
- Zeng, T.; Yu, X.; Chen, Z. Applying Artificial Intelligence in the Microbiome for Gastrointestinal Diseases: A review. J. Gastroenterol. Hepatol. 2021, 36, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Gubatan, J.; Levitte, S.; Patel, A.; Balabanis, T.; Wei, M.T.; Sinha, S.R. Artificial Intelligence Applications in Inflammatory Bowel Disease: Emerging Technologies and Future Directions. World J. Gastroenterol. 2021, 27, 1920–1935. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhang, X.; Hu, Z. Application of Artificial Intelligence Modeling Technology Based on Multi-Omics in Noninvasive Diagnosis of Inflammatory Bowel Disease. J. Inflamm. Res. 2021, 14, 1933–1943. [Google Scholar] [CrossRef]
- Ezziane, Z. Applications of Artificial Intelligence in Bioinformatics: A Review. Expert Syst. Appl. 2006, 30, 2–10. [Google Scholar] [CrossRef]
- Zucker, J.-D.; Clément, K. Artificial Intelligence at the Service of Metabolic Diseases. Med. Mal. Metab. 2021, 15, 70–79. [Google Scholar] [CrossRef]
- Smith, K.P.; Wang, H.; Durant, T.J.; Mathison, B.A.; Sharp, S.E.; Kirby, J.E.; Long, S.W.; Rhoads, D.D. Applications of Artificial Intelligence in Clinical Microbiology Diagnostic Testing. Clin. Microbiol. Newsl. 2020, 42, 61–70. [Google Scholar] [CrossRef]
- Benjamin, M.; Aisen, A.; Benjamin, E. Accelerating Development and Clinical Deployment of Diagnostic Im-aging Artificial Intelligence. J. Am. Coll. Radiol. 2021, 18, 1514–1516. [Google Scholar] [CrossRef] [PubMed]
- Wani, A.K.; Roy, P.; Kumar, V.; Mir, T.U.G. Metagenomics and Artificial Intelligence in the Context of Human Health. Infect. Genet. Evol. 2022, 100, 105267. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Han, K. Methodologic Guide for Evaluating Clinical Performance and Effect of Artificial Intelligence Technology for Medical Diagnosis and Prediction. Radiology 2018, 286, 800–809. [Google Scholar] [CrossRef]
- Prifti, E.; Chevaleyre, Y.; Hanczar, B.; Belda, E.; Danchin, A.; Clément, K.; Zucker, J.-D. Interpretable and Accurate Prediction Models for Metagenomics Data. GigaScience 2020, 9, giaa010. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xiao, Y.; Olsen, R.H.; Wang, C.; Meng, H.; Shi, L. Short- and Long-Read Metagenomics Insight into the Genetic Contexts and Hosts of Mobile Antibiotic Resistome in Chinese Swine Farms. Sci. Total Environ. 2022, 827, 154352. [Google Scholar] [CrossRef] [PubMed]
- Flygare, S.; Simmon, K.; Miller, C.; Qiao, Y.; Kennedy, B.; Di Sera, T.; Graf, E.H.; Tardif, K.D.; Kapusta, A.; Rynearson, S.; et al. Taxonomer: An Interactive Metagenomics Analysis Portal for Universal Pathogen Detection and Host mRNA Expression Profiling. Genome Biol. 2016, 17, 111. [Google Scholar] [CrossRef]


{kind=link}
| Sample ID | Miseq Genotypes | MinION Genotypes | ID of the MinION Hit | Reads/ Cluster | Consensus Length | Percent Identity | Fusion Protein Cleavage Site □ |
|---|---|---|---|---|---|---|---|
| 44 | VIIi | VIIi | chicken/Pakistan/Wadana_Kasur/PNI_PF_(14F)/2015 | 200 | 734 | 100 | virulent |
| 45 | VIIi II | VIIi II | chicken/Pakistan/Wadana_Kasur/PNI_PF_(14F)/2015 chicken/USA/LaSota/1946 | 28 5 | 734 733 | 99.31 96.44 | Virulent Low virulent |
| 46 | VIIi II | VIIi II | chicken/Pakistan/Wadana_Kasur/PNI_PF_(14F)/2015 chicken/USA/LaSota/1946 | 10 17 | 733 734 | 99.13 98.51 | Virulent Low virulent |
| 47 | VIIi II | ND d II | NA e chicken/USA/LaSota/1946 | NA 139 | NA 732 | NA 99.32 | NA Low virulent |
| 48 | ND VIIi | II VIIi | chicken/USA/LaSota/1946 chicken/Pakistan/Wadana_Kasur/PNI_PF_(14F)v/2015 | 200 21 | 732 733 | 99.59 99.13 | Low virulent virulent |
| 49 | VIIi II | ND II | NA chicken/USA/LaSota/1946 | NA 200 | NA 732 | NA 99.32 | NA Low virulent |
| 50 | VIIi | VIIi | chicken/Pakistan/Wadana_Kasur/PNI_PF_(14F)/2015 | 113 | 734 | 100 | virulent |
| 51 | VIIi | VIIi | chicken/Pakistan/Mirpur_Khas/3EOS/2015 | 200 | 734 | 100 | virulent |
| 52 a | VIIi | ND | NA | NA | NA | NA | NA |
| 53 | VIIi | VIIi | exotic Parakeets/Pakistan/Charah/Pk29/29A/2015 | 5 | 726 | 98.5 | virulent |
| 54 | NO NDV | NO NDV | NA | NA | NA | NA | NA |
| 55 | NO NDV | NO NDV | NA | NA | NA | NA | NA |
| 56 | NO NDV | NO NDV | NA | NA | NA | NA | NA |
| 57 | NO NDV | NO NDV | NA | NA | NA | NA | NA |
| 58 | VIIi | VIIi | chicken/Pakistan/Gharoo/Three_star_PF_(7G)/2015 | 8 | 729 | 99.32 | virulent |
| TN b | NA | ND | NA | NA | NA | NA | NA |
| EN c | NA | ND | NA | NA | NA | NA | NA |
| 1. Always use “standard operating procedures” for sample-to-sample comparison purposes. |
| 2. Develop “a priori” a sampling strategy focused on the specific problem with the help of a field veterinarian and pathologist. |
| 3. Develop a sampling strategy that covers “completely and evenly” the areas or the host of interest. |
| 4. Minimize contamination from operators, non-target tissues, and from the environment at all stages of collection. |
| 5. Minimize post-sampling contamination; use masks, sterile plasticware, media, and antibiotics if possible for manipulation and storage. |
| 6. Do not mix different types of samples (e.g., cloacal samples will dilute respiratory samples with bacterial nucleic acids). |
| 7. Obtain sufficient starting sample material (RNA/DNA) to minimize the amplification steps (e.g., pool the same type of samples if necessary). |
| 8. Minimize degradation of nucleic acids (RNAs are very sensitive) by using gloves, cold chains, and RNAse-free reagents. |
| 9. Use trained operators at all stages of the process. |
| 10.Use fast and reliable labeling (printed tags, barcoding, spreadsheets, instead of pens at the site). |
| 11. Obtain and link the most complete metadata possible in all samples (e.g., farm clinical and management information). |
| 12. Note “all’ clinical details associated with the host pathology for each individual sample. |
| 13. When spotting on FTA cards, rigorously follow the recommendations on expiration dates, spotting volumes, drying time storage, and shipment conditions. |
| 14. Include information in “the shipping form” that will be used for the interpretation of complex results such as: |
| Date of collection, the name of the operator, and/or sample contact information. |
| Flock identification (can be coded for confidentiality) |
| Type of sample (oropharyngeal, cloacal, tissue). |
| Species and age of the sampled birds. |
| Optional information: vaccination; suspected disease; clinical lesions; histology; flock health; production problems; GPS location. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonso, C.L.; Afonso, A.M. Next-Generation Sequencing for the Detection of Microbial Agents in Avian Clinical Samples. Vet. Sci. 2023, 10, 690. https://doi.org/10.3390/vetsci10120690
Afonso CL, Afonso AM. Next-Generation Sequencing for the Detection of Microbial Agents in Avian Clinical Samples. Veterinary Sciences. 2023; 10(12):690. https://doi.org/10.3390/vetsci10120690
Chicago/Turabian StyleAfonso, Claudio L., and Anna M. Afonso. 2023. "Next-Generation Sequencing for the Detection of Microbial Agents in Avian Clinical Samples" Veterinary Sciences 10, no. 12: 690. https://doi.org/10.3390/vetsci10120690