
Efficient Robust Yield Method for Preparing Bacterial Ghosts by Escherichia coli Phage ID52 Lysis Protein E


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, and Culture Conditions
2.2. Lysis Protein E Expressing Plasmid Construction
2.3. Induction Expression of Lysis Protein E
2.4. Scanning Electron Microscopy (SEM)
2.5. Transmission Electron Microscopy (TEM)
2.6. Western Blot Analysis
2.7. Construction of EcN with araBAD Deletion Mutation and ST with araBAD Deletion Mutation
2.8. Statistical Analysis
3. Results
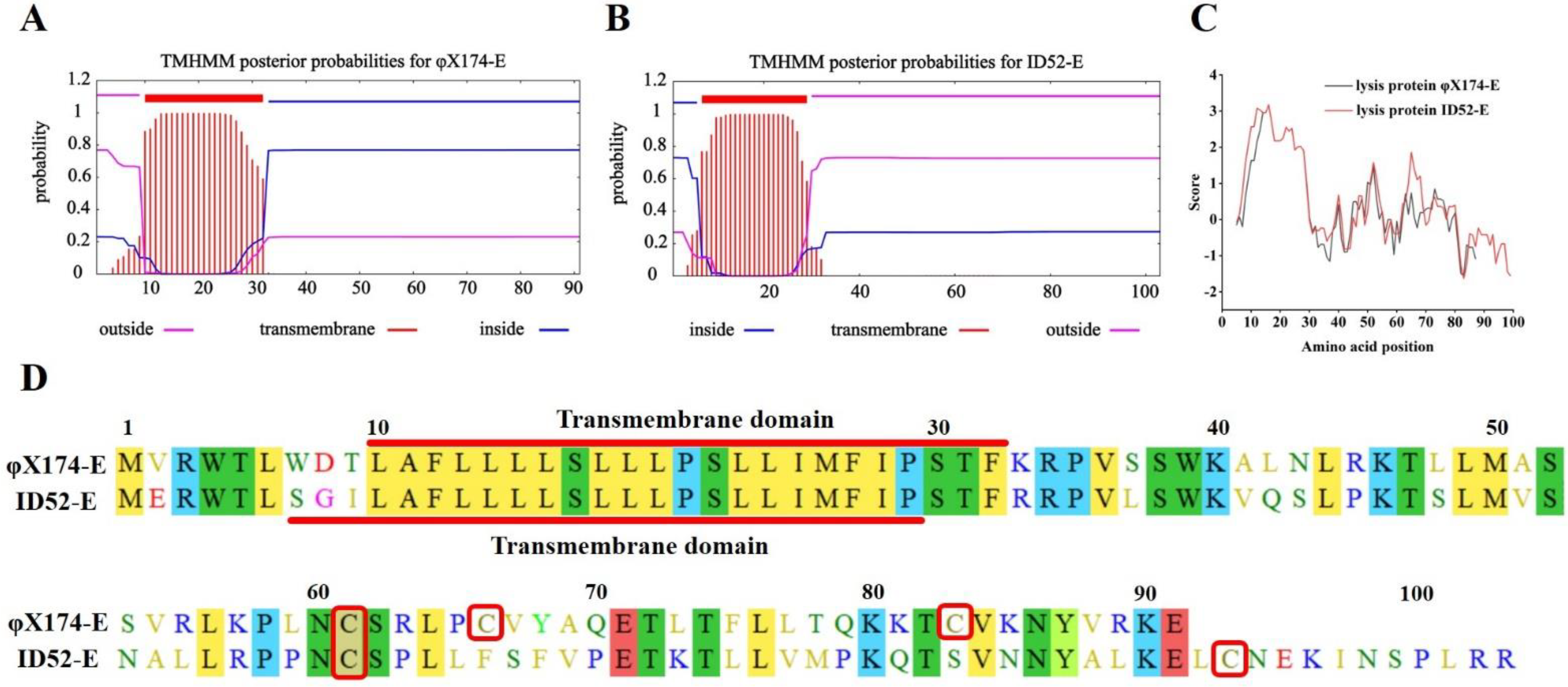
3.1. Lysis Protein E Expressing Plasmid Construction
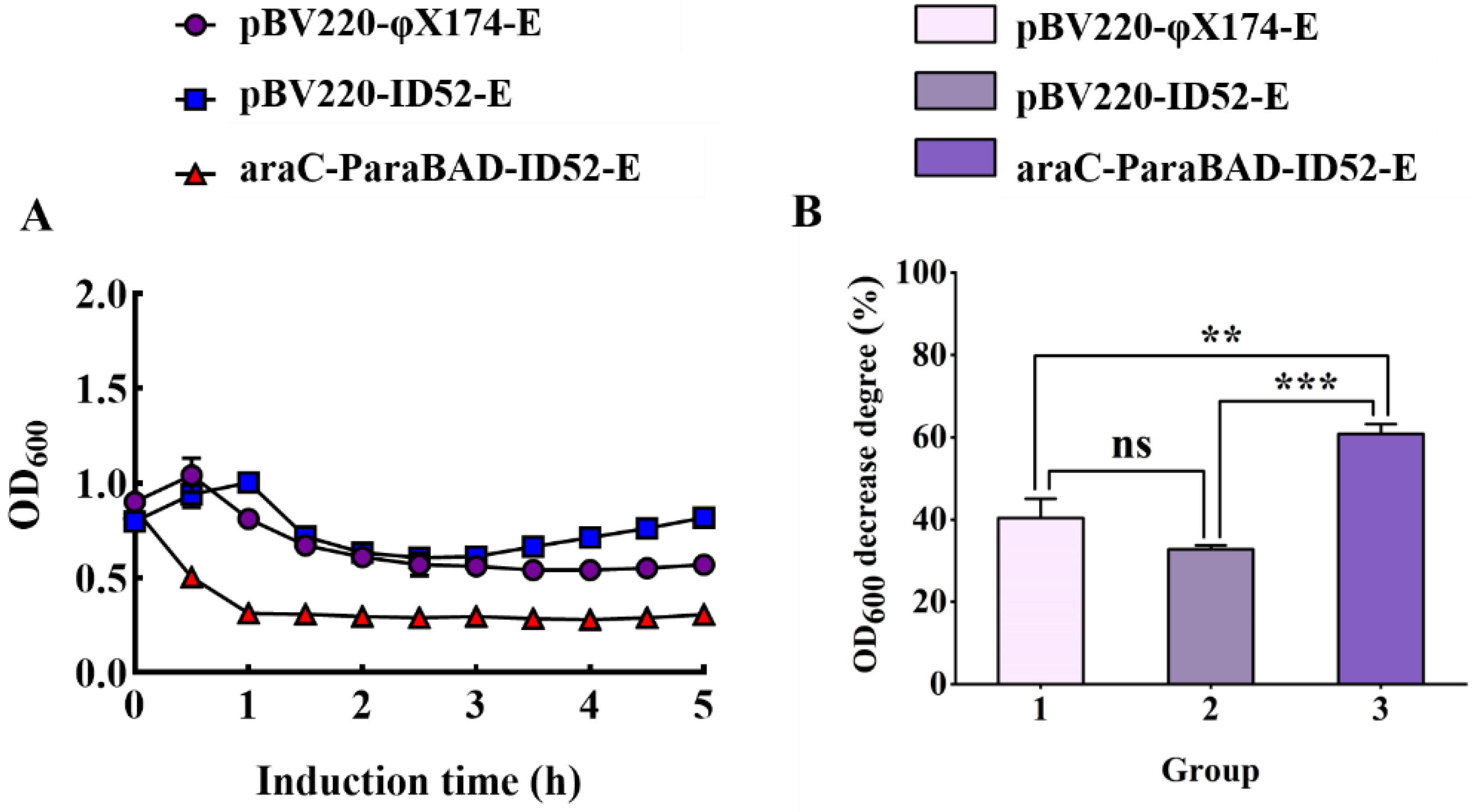
3.2. Lysis Activity of Protein φ174-E and ID52-E in E. coli BL21(DE3)
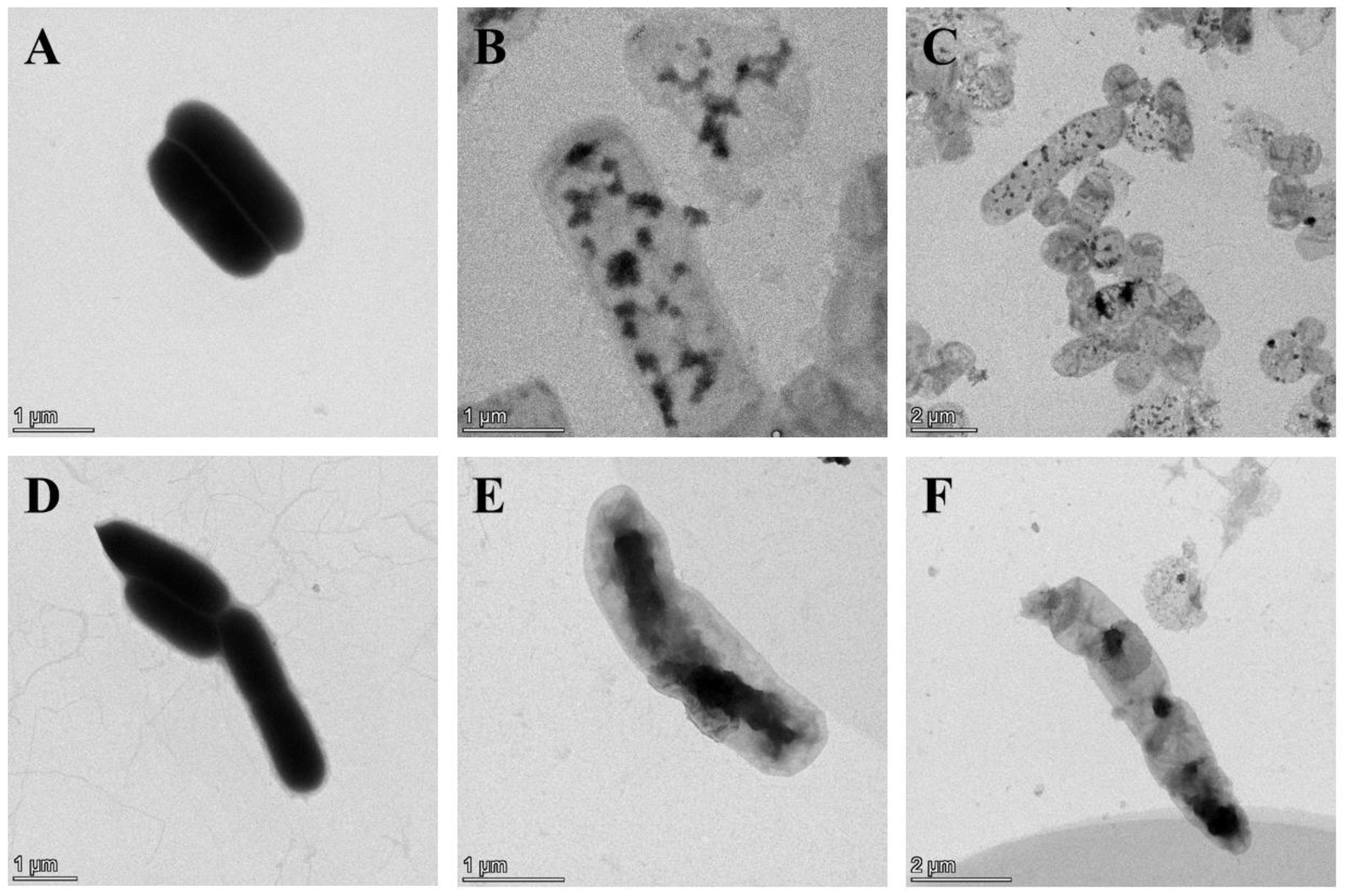
3.3. Morphological Observation of E. coli BL21(DE3) BGs by SEM and TEM
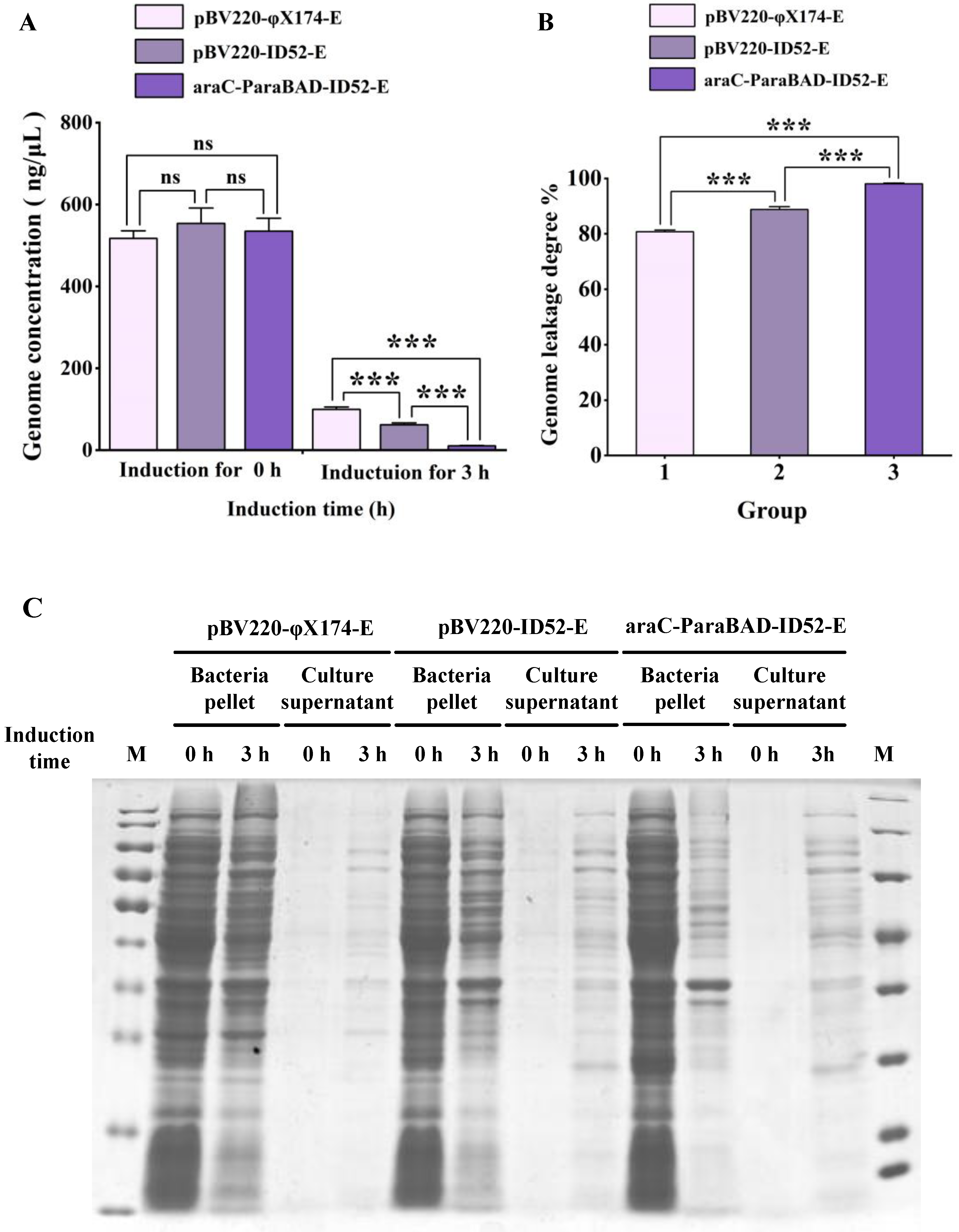
3.4. Detecting the Expression Level and Expression Location of Lysis Protein E by Western Blot
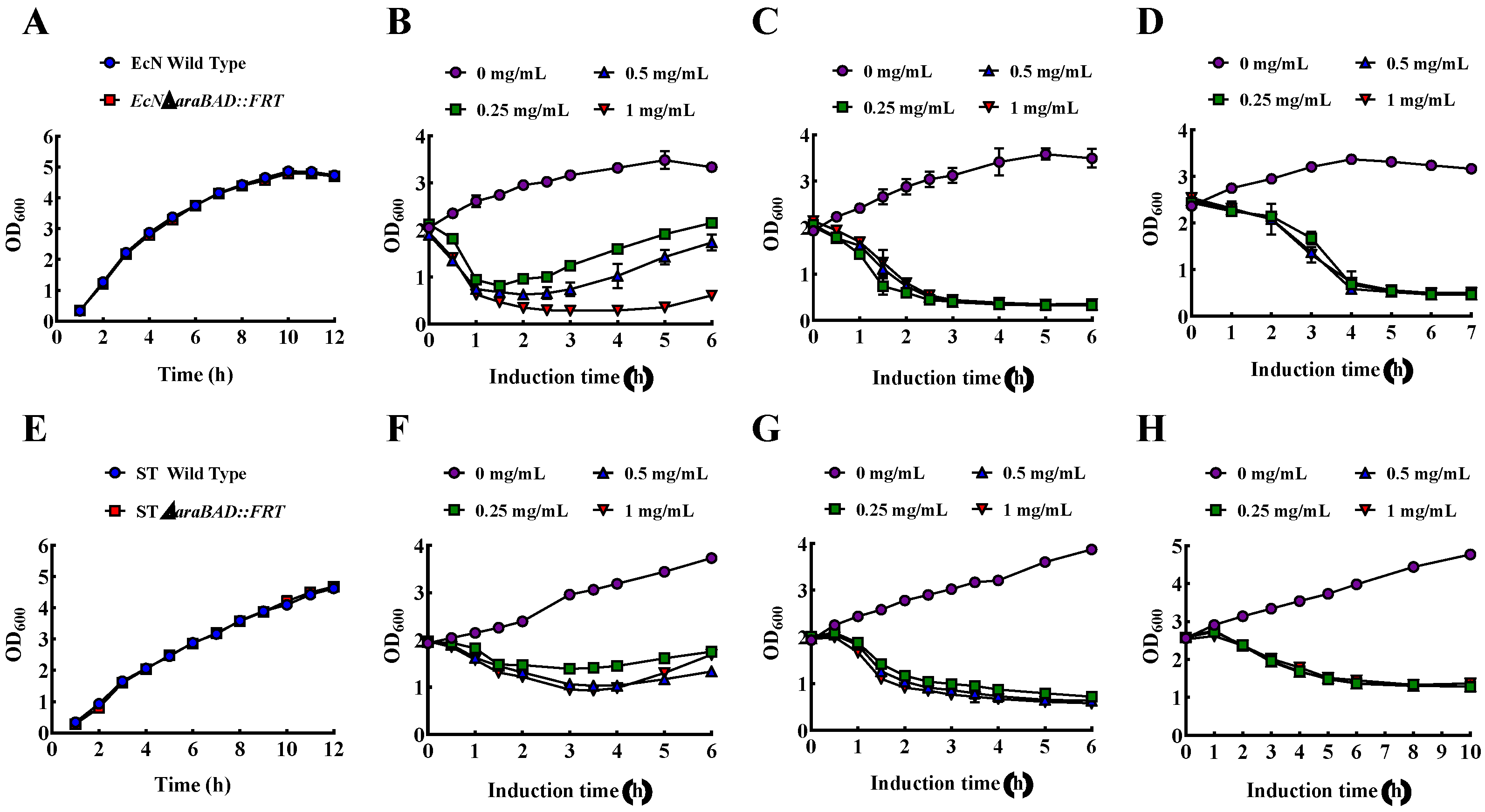
3.5. Construction of Engineered Strain EcN with araBAD Deletion Mutation and ST with araBAD Deletion Mutation
3.6. Growth, Lysis, and Characterization of EcN BGs and ST BGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Langemann, T.; Koller, V.J.; Muhammad, A.; Kudela, P.; Mayr, U.B.; Lubitz, W. The Bacterial Ghost platform system: Production and applications. Bioeng. Bugs 2010, 1, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganeshpurkar, A.; Ganeshpurkar, A.; Pandey, V.; Agnihotri, A.; Bansal, D.; Dubey, N. Harnessing the potential of bacterial ghost for the effective delivery of drugs and biotherapeutics. Int. J. Pharm. Investig. 2014, 4, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abtin, A.; Kudela, P.; Mayr, U.B.; Koller, V.J.; Mildner, M.; Tschachler, E.; Lubitz, W. Escherichia coli ghosts promote innate immune responses in human keratinocytes. Biochem. Biophys. Res. Commun. 2010, 400, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Tu, F.P.; Chu, W.H.; Zhuang, X.Y.; Lu, C.P. Effect of oral immunization with Aeromonas hydrophila ghosts on protection against experimental fish infection. Lett. Appl. Microbiol. 2010, 50, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Riedmann, E.M.; Kyd, J.M.; Cripps, A.W.; Lubitz, W. Bacterial ghosts as adjuvant particles. Expert Rev. Vaccines 2007, 6, 241–253. [Google Scholar] [CrossRef]
- Peng, W.; Si, W.; Yin, L.; Liu, H.F.; Yu, S.Y.; Liu, S.G.; Wang, C.L.; Chang, Y.H.; Zhang, Z.; Hu, S.P.; et al. Salmonella enteritidis ghost vaccine induces effective protection against lethal challenge in specific-pathogen-free chicks. Immunobiology 2011, 216, 558–565. [Google Scholar] [CrossRef]
- Di Pasquale, A.; Preiss, S.; Tavares Da Silva, F.; Garcon, N. Vaccine Adjuvants: From 1920 to 2015 and Beyond. Vaccines 2015, 3, 320–343. [Google Scholar] [CrossRef] [Green Version]
- Kudela, P.; Koller, V.J.; Lubitz, W. Bacterial ghosts (BGs)—Advanced antigen and drug delivery system. Vaccine 2010, 28, 5760–5767. [Google Scholar] [CrossRef]
- Hu, J.; Zuo, J.; Chen, Z.; Fu, L.; Lv, X.; Hu, S.; Shi, X.; Jing, Y.; Wang, Y.; Wang, Z.; et al. Use of a modified bacterial ghost lysis system for the construction of an inactivated avian pathogenic Escherichia coli vaccine candidate. Vet. Microbiol. 2019, 229, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Jechlinger, W.; Haller, C.; Resch, S.; Hofmann, A.; Szostak, M.P.; Lubitz, W. Comparative immunogenicity of the hepatitis B virus core 149 antigen displayed on the inner and outer membrane of bacterial ghosts. Vaccine 2005, 23, 3609–3617. [Google Scholar] [CrossRef]
- Gong, S.S.; Nan, N.; Sun, Y.K.; He, Z.L.; Li, J.J.; Chen, F.H.; Li, T.; Ning, N.Z.; Wang, J.X.; Li, Z.; et al. Protective Immunity Elicited by VP1 Chimeric Antigens of Bacterial Ghosts against Hand-Foot-and-Mouth Disease Virus. Vaccines 2020, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paukner, S.; Kohl, G.; Jalava, K.; Lubitz, W. Sealed bacterial ghosts--novel targeting vehicles for advanced drug delivery of water-soluble substances. J. Drug Target 2003, 11, 151–161. [Google Scholar] [PubMed]
- Paukner, S.; Kohl, G.; Lubitz, W. Bacterial ghosts as novel advanced drug delivery systems: Antiproliferative activity of loaded doxorubicin in human Caco-2 cells. J. Control. Release 2004, 94, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Ebensen, T.; Paukner, S.; Link, C.; Kudela, P.; de Domenico, C.; Lubitz, W.; Guzman, C.A. Bacterial ghosts are an efficient delivery system for DNA vaccines. J. Immunol. 2004, 172, 6858–6865. [Google Scholar] [CrossRef] [Green Version]
- Lubitz, W.; Witte, A.; Eko, F.O.; Kamal, M.; Jechlinger, W.; Brand, E.; Marchart, J.; Haidinger, W.; Huter, V.; Felnerova, D.; et al. Extended recombinant bacterial ghost system. J. Biotechnol. 1999, 73, 261–273. [Google Scholar] [CrossRef]
- Paukner, S.; Stiedl, T.; Kudela, P.; Bizik, J.; Al Laham, F.; Lubitz, W. Bacterial ghosts as a novel advanced targeting system for drug and DNA delivery. Expert Opin. 2006, 3, 11–22. [Google Scholar] [CrossRef]
- Mayr, U.B.; Walcher, P.; Azimpour, C.; Riedmann, E.; Haller, C.; Lubitz, W. Bacterial ghosts as antigen delivery vehicles. Adv. Drug Deliv. Rev. 2005, 57, 1381–1391. [Google Scholar] [CrossRef]
- Batah, A.M.; Ahmad, T.A. The development of ghost vaccines trials. Expert Rev. Vaccines 2020, 19, 549–562. [Google Scholar] [CrossRef]
- Witte, A.; Wanner, G.; Blasi, U.; Halfmann, G.; Szostak, M.; Lubitz, W. Endogenous Transmembrane Tunnel Formation Mediated by pX174 Lysis Protein E. J. Bacteriol. 1990, 172, 4109–4114. [Google Scholar] [CrossRef] [Green Version]
- Witte, A.; Wanner, G.; Sulzner, M.; Lubitz, W. Dynamics of PhiX174 protein E-mediated lysis of Escherichia coli. Arch. Microbiol. 1992, 157, 381–388. [Google Scholar] [CrossRef]
- Witte, A.; Brand, E.; Mayrhofer, P.; Narendja, F.; Lubitz, W. Mutations in cell division proteins FtsZ and FtsA inhibit φX174 protein-E-mediated lysis of Escherichia coli. Arch. Microbiol. 1998, 170, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.X.; Zhang, Y.Y.; Liu, X.L. Efficient production of safety-enhanced Escherichia coli ghosts by tandem expression of PhiX 174 mutant gene E and staphylococcal nuclease A gene. Microbiol. Res. 2015, 176, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Peng, W.; Si, W.; Yin, L.; Liu, S.G.; Liu, H.F.; Zhao, H.L.; Wang, C.L.; Chang, Y.H.; Lin, Y.Z. Enhancement of bacteriolysis of Shuffled phage PhiX174 gene E. Virol. J. 2011, 8, 206. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Cui, L.; Wang, M.; Sun, Q.; Liu, K.; Wang, J. A Novel and Efficient High-Yield Method for Preparing Bacterial Ghosts. Toxins 2021, 13, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.A.; Salem-Bekhit, M.M.; Alanazi, F.K. Sponge-like: A new protocol for preparing bacterial ghosts. Sci. World J. 2013, 2013, 545741–545747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Oh, S.; Vinod, N.; Ji, S.M.; Noh, H.B.; Koo, J.M.; Lee, S.H.; Kim, S.C.; Lee, K.S.; Choi, C.W. Characterization of Chemically-Induced Bacterial Ghosts (BGs) Using Sodium Hydroxide-Induced Vibrio parahaemolyticus Ghosts (VPGs). Int. J. Mol. Sci. 2016, 17, 1904–1918. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Hao, L.; Liu, X.; Borras-Hidalgo, O.; Zhang, Y. Enhanced anti-proliferative efficacy of epothilone B loaded with Escherichia coli Nissle 1917 bacterial ghosts on the HeLa cells by mitochondrial pathway of apoptosis. Drug Dev. Ind. Pharm. 2018, 44, 1328–1335. [Google Scholar] [CrossRef]
- Herrero-Fresno, A.; Olsen, J.E. Salmonella Typhimurium metabolism affects virulence in the host—A mini-review. Food Microbiol. 2018, 71, 98–110. [Google Scholar] [CrossRef]
- Jawale, C.V.; Lee, J.H. Evaluation of immunogenicity and protective efficacy of adjuvanted Salmonella Typhimurium ghost vaccine against salmonellosis in chickens. Vet. Q. 2016, 36, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Rokyta, D.R.; Burch, C.L.; Caudle, S.B.; Wichman, H.A. Horizontal gene transfer and the evolution of microvirid coliphage genomes. J. Bacteriol. 2006, 188, 1134–1142. [Google Scholar] [CrossRef] [Green Version]
- van den Ent, F.; Lowe, J. RF cloning: A restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 2006, 67, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Bond, S.R.; Naus, C.C. RF-Cloning.org: An online tool for the design of restriction-free cloning projects. Nucleic Acids Res. 2012, 40, W209–W213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mykytczuk, N.C.; Trevors, J.T.; Leduc, L.G.; Ferroni, G.D. Fluorescence polarization in studies of bacterial cytoplasmic membrane fluidity under environmental stress. Prog. Biophys. Mol. Biol. 2007, 95, 60–82. [Google Scholar] [CrossRef] [PubMed]
- Gubellini, F.; Verdon, G.; Karpowich, N.K.; Luff, J.D.; Boel, G.; Gauthier, N.; Handelman, S.K.; Ades, S.E.; Hunt, J.F. Physiological Response to Membrane Protein Overexpression in E. coli. Mol. Cell. Proteom. 2011, 10, M111.007930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuller, A.; Harkness, R.E.; Ruther, U.; Lubitzl, W. Deletion of C-terminal amino acid codons of PhiX174 gene E: Effect on its lysis inducing properties. Nucleic Acids Res. 1985, 13, 4143–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maratea, D.; Young, K.; Young, R. Deletion and fusion analysis of the phage φX174 lysis gene E. Gene 1985, 40, 39–46. [Google Scholar] [CrossRef]
- Bläsi, U.; Lubitz, W. Influence of C-terminal Modifications of φX174 Lysis Gene E on its Lysis inducing Properties. J. Gen. Virol. 1985, 66, 1209–1213. [Google Scholar] [CrossRef]
- Bläsi, U.; Linke, R.P.; Lubitz, W. Evidence for membrane-bound oligomerization of bacteriophage ϕ X174 lysis protein-E. J. Biol. Chem. 1989, 264, 4552–4558. [Google Scholar] [CrossRef]
- Bläsi, U.; Linke, R.P.; Lubitz, W. Proline 21, a residue within the alpha-helical domain of phiX174 lysis protein E, is required for its function in Escherichia coli. Biol. Chem. 1989, 264, 2552–2558. [Google Scholar]
- Ma, J.; Bruce, T.J.; Jones, E.M.; Cain, K.D. A Review of Fish Vaccine Development Strategies: Conventional Methods and Modern Biotechnological Approaches. Microorganisms 2019, 7, 569. [Google Scholar] [CrossRef] [Green Version]
- Koller, V.J.; Dirsch, V.M.; Beres, H.; Donath, O.; Reznicek, G.; Lubitz, W.; Kudela, P. Modulation of bacterial ghosts—induced nitric oxide production in macrophages by bacterial ghost-delivered resveratrol. FEBS J. 2013, 280, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Paukner, S.; Kudela, P.; Kohl, G.; Schlapp, T.; Friedrichs, S.; Lubitz, W. DNA-loaded bacterial ghosts efficiently mediate reporter gene transfer and expression in macrophages. Mol. Ther. 2005, 11, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Hatfaludi, T.; Liska, M.; Zellinger, D.; Ousman, J.P.; Szostak, M.; Ambrus, A.; Jalava, K.; Lubitz, W. Bacterial ghost technology for pesticide delivery. J. Agric. Food Chem. 2004, 52, 5627–5634. [Google Scholar] [CrossRef] [PubMed]
- Walcher, P.; Cui, X.L.; Arrow, J.A.; Scobie, S.; Molinia, F.C.; Cowan, P.E.; Lubitz, W.; Duckworth, J.A. Bacterial ghosts as a delivery system for zona pellucida-2 fertility control vaccines for brushtail possums (Trichosurus vulpecula). Vaccine 2008, 26, 6832–6838. [Google Scholar] [CrossRef]
- Doore, S.M.; Fane, B.A. The microviridae: Diversity, assembly, and experimental evolution. Virology 2016, 491, 45–55. [Google Scholar] [CrossRef]
- Chamakura, K.R.; Young, R. Single-gene lysis in the metagenomic era. Curr. Opin. Microbiol. 2020, 56, 109–117. [Google Scholar] [CrossRef]
- Cahill, J.; Young, R. Phage Lysis: Multiple Genes for Multiple Barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar]
- Wadle, D.; Henrich, B.; Plapp, R. Effects of mutations in genes fadR, fabB, fadE, and envC of Escherichia coli on the action of the lysis gene of bacteriophage φ X174. Curr. Microbiol. 1986, 14, 65–69. [Google Scholar] [CrossRef]
- Dvorak, P.; Chrast, L.; Nikel, P.I.; Fedr, R.; Soucek, K.; Sedlackova, M.; Chaloupkova, R.; de Lorenzo, V.; Prokop, Z.; Damborsky, J. Exacerbation of substrate toxicity by IPTG in Escherichia coli BL21(DE3) carrying a synthetic metabolic pathway. Microb. Cell Fact. 2015, 14, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Zhang, P.; Zhang, Z.; Liu, Y.; Chen, M.; Li, S.; Li, X. Bacterial navigation for tumor targeting and photothermally-triggered bacterial ghost transformation for spatiotemporal drug release. Acta Biomater. 2021, 131, 172–184. [Google Scholar] [CrossRef]
- Ye, S.; Kim, J.W.; Kim, S.R. Metabolic Engineering for Improved Fermentation of L-Arabinose. J. Microbiol. Biotechnol. 2019, 29, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains Plasmids Primers | Description | Source |
|---|---|---|
| Strains | ||
| DH5α | Host cells for plasmid amplification | TIANGEN BIOTECH, Beijing, China |
| BL21(DE3) | Host cells for protein expression | TIANGEN BIOTECH, Beijing, China |
| E. coli Nissle 1917 | Wild type | Mutaflor, Germany |
| E. coli Nissle 1917 ΔaraBAD::FRT | Deletion of araBAD gene and insertion of FRT locusin E. coli Nissle 1917 | This study |
| ST | Wild type of Salmonella enterica subsp. enterica serovarTyphimurium str. ATCC 14028 | Institute of Microbiology, Guangdong Academy of Science, China |
| ST ΔaraBAD::FRT | Deletion of araBAD gene and insertion of FRT locus in ST | This study |
| Plasmids | ||
| pBV220-sGFP-Chl | The template of constructing expression vector of lysis gene E | Our lab |
| pET29a-φX174-E | The template of constructing expression vector ofgene φX174-E | Our lab |
| pET29a-ID52-E | The template of constructing expression vector of gene ID52-E | Our lab |
| pBV220-φX174-E | Lysis plasmid used in this study | This study |
| pBV220-ID52-E | Lysis plasmid used in this study | This study |
| araC-ParaBAD-ID52-E | Lysis plasmid used in this study | This study |
| pKD4 | Plasmid for λ Red homologous recombination | Our lab |
| pKD46 | Plasmid for λ Red homologous recombination | Our lab |
| pCP20 | Plasmid for λ Red homologous recombination | Our lab |
| Primers | ||
| φX174-E-F | TTGGTTAAAAATTAAGGAGGAATTCATGGTACGCTGGACTTTGTG | |
| φX174-E-R | ACAGCCAAGCTTGGCTGCAGTTATTTTTCAAACTGCGGATG | |
| ID52-E-F | TAAAAATTAAGGAGGAATTCATGGAACGCTGGACCTTAAG | |
| ID52-E-R | ACAGCCAAGCTTGGCTGCAGTTATTTTTCAAACTGCGGATG | |
| araC-ParaBAD-F | TGCGCCGACCAGAACACCTTGCCGATTATGACAACTTGACGGCTACA | |
| araC-ParaBAD-R | TGCCGCTTAAGGTCCAGCGTTCCATTTTTTATAACCTCCTTAGAGCTCG | |
| linearized pBV220-ID52-E-F | ATGGAACGCTGGACCTTAAG | |
| linearized pBV220-ID52-E-R | TCGGCAAGGTGTTCTGGT | |
| P1 | ATGACACCGGACATTATCCTG | |
| P2 | GTGCTTTCAGTGGATTTCGG | |
| P3 | TTTTTCGCAACTCTCTACTGTTTCTCCATACCCGTTTTTTTGGATGGAGTGAAACGGTGTAGGCTGGAGCTGCTTC | |
| P4 | CTGGTTTCGTTCCAAAACCAAAATTTATTTTGATTGGCTGTGGTTTTATACAGTCACATATGAATATCCTCCTTAG | |
| P5 | TTTGCCGCGACTCTCTACTGTTTCTCCATACCTGTTTTTCTGGATGGAGTAAGACGGTGTAGGCTGGAGCTGCTTC | |
| P6 | TATATCACCGACCAGATTCATCAACGCGCCCCCCATGGGAGCGTTTTTAGAGGCACATATGAATATCCTCCTTAG | |
| P7 | TTAGCGGATCCAGCCTGA | |
| P8 | TGCAGCATTCGCAGATCG | |
| P9 | GATTAGCGGATCCTGCCTGA | |
| P10 | TATCAAAGCGCATTTGCTGAA | |
| P11 | AAGGGATAAATATCTAACACCGTGC | |
| P12 | ACGGCATAGTGCGTGTTTATG | |
| Initial Induction OD600 Values | Lysis Efficiency (%) | ||
|---|---|---|---|
| pBV220-φX174-E | pBV220-ID52-E | araC-ParaBAD- ID52-E | |
| 0.8 | 100 | 99.984 | 99.755 |
| 1.2 | 100 | 99.994 | 99.994 |
| 1.6 | 100 | 99.998 | 99.997 |
| 2.0 | 100 | 100 | 99.998 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Zhu, W.; Zhu, G.; Xu, Y.; Li, S.; Chen, R.; Chen, L.; Wang, J. Efficient Robust Yield Method for Preparing Bacterial Ghosts by Escherichia coli Phage ID52 Lysis Protein E. Bioengineering 2022, 9, 300. https://doi.org/10.3390/bioengineering9070300
Ma Y, Zhu W, Zhu G, Xu Y, Li S, Chen R, Chen L, Wang J. Efficient Robust Yield Method for Preparing Bacterial Ghosts by Escherichia coli Phage ID52 Lysis Protein E. Bioengineering. 2022; 9(7):300. https://doi.org/10.3390/bioengineering9070300
Chicago/Turabian StyleMa, Yi, Wenjun Zhu, Guanshu Zhu, Yue Xu, Shuyu Li, Rui Chen, Lidan Chen, and Jufang Wang. 2022. "Efficient Robust Yield Method for Preparing Bacterial Ghosts by Escherichia coli Phage ID52 Lysis Protein E" Bioengineering 9, no. 7: 300. https://doi.org/10.3390/bioengineering9070300