
Process- and Product-Related Foulants in Virus Filtration


Abstract
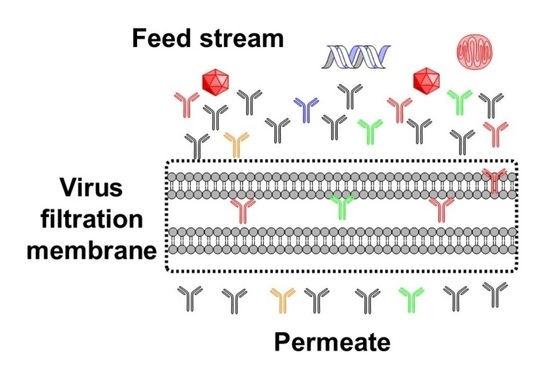
:
1. Introduction
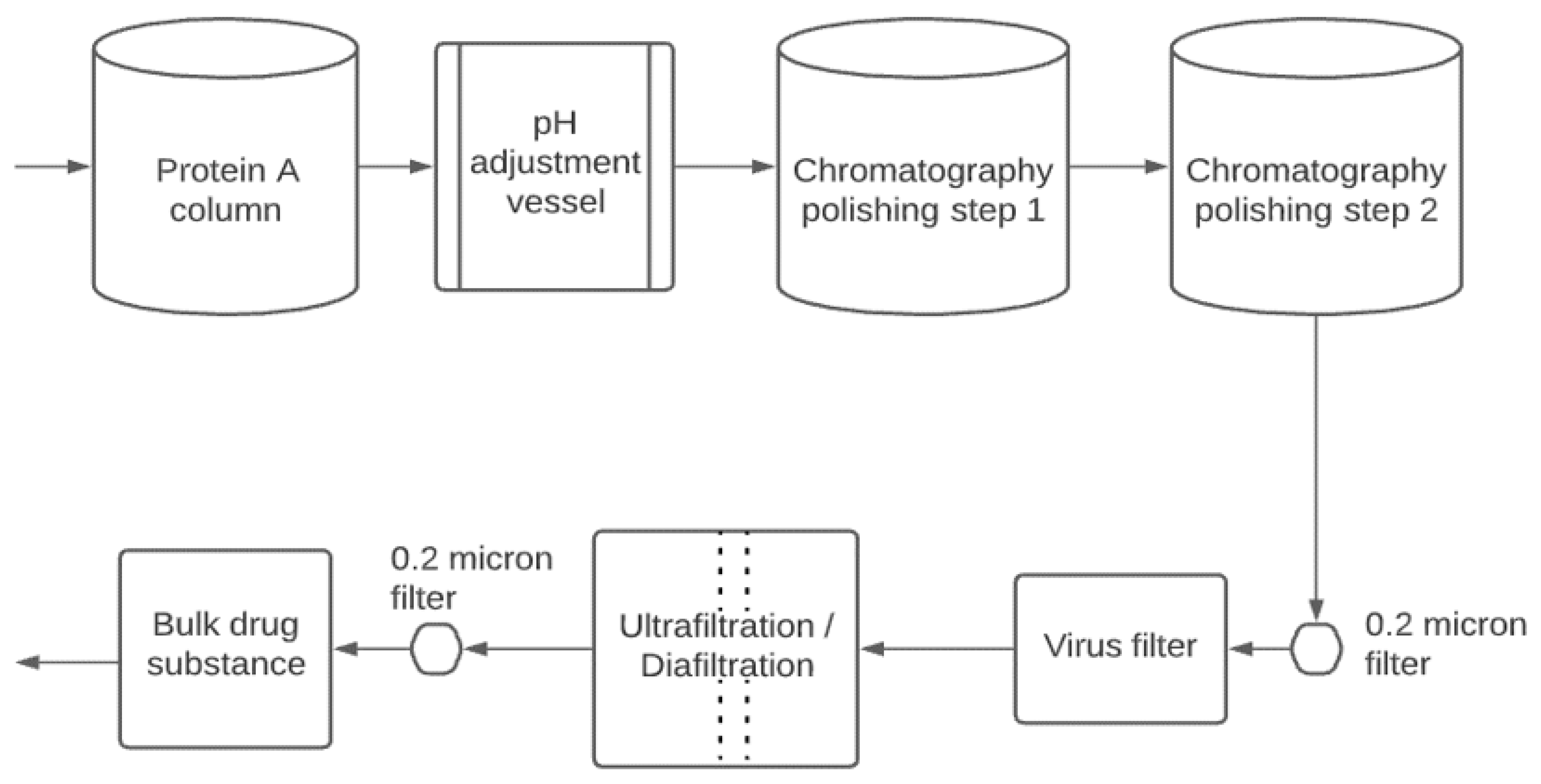
2. Downstream Processing
2.1. Platform Processes
2.2. Viruses, Virus Clearance, and Virus Filters
3. Virus Filter Foulants
3.1. Monoclonal Antibody Aggregates
3.1.1. Reversible Aggregates
3.1.2. Irreversible Aggregates
3.2. Host Cell Proteins (HCP), Proteases, and Nucleic Acids
3.3. Endotoxins
3.4. Product-Mediated Foulants
3.4.1. Charge Variants
3.4.2. Denatured Variants
3.4.3. Sequence Variants
3.4.4. Micro-Heterogeneity-Induced Product Variants
4. Mitigation of Virus Filter Fouling
4.1. Prefiltration before Virus Filtration
4.2. Mitigation of Virus Filter Fouling Using Process Parameters
5. Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Troccoli, N.M.; McIver, J.; Losikoff, A.; Poiley, J. Removal of viruses from human intravenous immune globulin by 35 nm nanofiltration. Biologicals 1998, 26, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.R.; Namila; Fan, R.; Qian, X. Virus Removal and Virus Purification. In Current Trends and Future Developments on (Bio-) Membranes; Elsevier: Amsterdam, The Netherlands, 2019; pp. 69–96. [Google Scholar] [CrossRef]
- FDA. Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use; Bethesda: Rockville, MD, USA, 1997.
- Han, B.; Carlson, J.O.; Powers, S.M.; Wickramasinghe, S.R. Enhanced virus removal by flocculation and microfiltration. Biotechnol. Bioprocess Eng. 2002, 7, 6–9. [Google Scholar] [CrossRef]
- Billups, M.; Minervini, M.; Holstein, M.; Feroz, H.; Ranjan, S.; Hung, J.; Bao, H.; Ghose, S.; Li, Z.J.; Zydney, A.L. Antibody retention by virus filtration membranes: Polarization and sieving effects. J. Membr. Sci. 2021, 620, 118884. [Google Scholar] [CrossRef]
- Dhara, V.G.; Naik, H.M.; Majewska, N.I.; Betenbaugh, M.J. Recombinant Antibody Production in CHO and NS0 Cells: Differences and Similarities. BioDrugs 2018, 32, 571–584. [Google Scholar] [CrossRef]
- Global Market Insights. Monoclonal Antibodies Market Size By Type (Fully Human, Humanized, Chimeric), By Application (Oncology, Autoimmune Diseases, Infectious Diseases), By End-use (Hospitals, Clinics), COVID-19 Impact Analysis, Regional Outlook, Application Potential, Competitive Market Share & Forecast, 2021–2027. Available online: https://www.gminsights.com/industry-analysis/monoclonal-antibodies-market (accessed on 19 March 2022).
- Wagner, E.; Colas, O.; Chenu, S.; Goyon, A.; Murisier, A.; Cianferani, S.; Francois, Y.; Fekete, S.; Guillarme, D.; D’Atri, V.; et al. Determination of size variants by CE-SDS for approved therapeutic antibodies: Key implications of subclasses and light chain specificities. J. Pharm. Biomed. Anal. 2020, 184, 113166. [Google Scholar] [CrossRef] [PubMed]
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human cell lines for biopharmaceutical manufacturing: History, status, and future perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Patel, P.; Strauss, D.; Qian, X.; Wickramasinghe, S.R. Modeling flux in tangential flow filtration using a reverse asymmetric membrane for Chinese hamster ovary cell clarification. Biotechnol. Prog. 2021, 37, e3115. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Leber, J.; Loewe, D.; Lothert, K.; Oppermann, T.; Zitzmann, J.; Weidner, T.; Salzig, D.; Wolff, M.; Czermak, P. Chapter 5—Purification of New Biologicals Using Membrane-Based Processes. In Current Trends and Future Developments on (Bio-) Membranes; Basile, A., Charcosset, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 123–150. [Google Scholar] [CrossRef]
- Wickramasinghe, S.R.; Stump, E.D.; Grzenia, D.L.; Husson, S.M.; Pellegrino, J. Understanding virus filtration membrane performance. J. Membr. Sci. 2010, 365, 160–169. [Google Scholar] [CrossRef]
- Zalai, D.; Kopp, J.; Kozma, B.; Küchler, M.; Herwig, C.; Kager, J. Microbial technologies for biotherapeutics production: Key tools for advanced biopharmaceutical process development and control. Drug Discov. Today Technol. 2020, 38, 9–24. [Google Scholar] [CrossRef] [PubMed]
- DePalma, A. Continuity Promotes Bioprocessing Intensity. Genet. Eng. Biotechnol. News 2016, 36, 1–24. [Google Scholar] [CrossRef]
- Vunnum, S.; Vedantham, G.; Hubbard, B. Protein A-Based Affinity Chromatography. In Process Scale Purification of Antibodies; Gottschalk, U., Ed.; Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 113–133. [Google Scholar] [CrossRef]
- Bramer, C.; Tunnermann, L.; Gonzalez Salcedo, A.; Reif, O.W.; Solle, D.; Scheper, T.; Beutel, S. Membrane Adsorber for the Fast Purification of a Monoclonal Antibody Using Protein A Chromatography. Membranes 2019, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Chollangi, S.; Parker, R.; Singh, N.; Li, Y.; Borys, M.; Li, Z. Development of robust antibody purification by optimizing protein-A chromatography in combination with precipitation methodologies. Biotechnol. Bioeng. 2015, 112, 2292–2304. [Google Scholar] [CrossRef]
- Huang, P.Y.; Peterson, J. Scaleup and virus clearance studies on virus filtration in monoclonal antibody manufacture. In Membrane Separations in Biotechnology; Marcel Dekker: New York, NY, USA, 2001. [Google Scholar]
- Bolton, G.R.; Spector, S.; LaCasse, D. Increasing the capacity of parvovirus-retentive membranes: Performance of the Viresolve™ Prefilter. Biotechnol. Appl. Biochem. 2006, 43, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Miesegaes, G.R.; Lute, S.C.; Read, E.K.; Brorson, K.A. Viral clearance by flow-through mode ion exchange columns and membrane adsorbers. Biotechnol. Prog. 2014, 30, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Burnouf, T.; Radosevich, M. Nanofiltration of plasma-derived biopharmaceutical products. Haemophilia 2003, 9, 24–37. [Google Scholar] [CrossRef]
- Wieser, A.; Berting, A.; Medek, C.; Poelsler, G.; Kreil, T.R. The evolution of down-scale virus filtration equipment for virus clearance studies. Biotechnol. Bioeng. 2015, 112, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Kern, G.; Krishnan, M. Virus Removal by Filtration: Points to Consider. BioPharm Int. 2006, 19, 32–41. [Google Scholar]
- Bieberbach, M.; Kosiol, P.; Seay, A.; Bennecke, M.; Hansmann, B.; Hepbildikler, S.; Thom, V. Investigation of fouling mechanisms of virus filters during the filtration of protein solutions using a high throughput filtration screening device. Biotechnol. Prog. 2019, 35, e2776. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Roush, D.; Cramer, S. Domain contributions to antibody retention in multimodal chromatography systems. J. Chromatogr. A 2018, 1563, 89–98. [Google Scholar] [CrossRef]
- Gefroh, E.; Dehghani, H.; McClure, M.; Connell-Crowley, L.; Vedantham, G. Use of MMV as a Single Worst-Case Model Virus in Viral Filter Validation Studies. PDA J. Pharm. Sci. Technol. 2014, 68, 297. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Chen, S.; Bolton, G.; Chen, Q.; Lute, S.; Fisher, J.; Brorson, K. Virus filtration: A review of current and future practices in bioprocessing. Biotechnol Bioeng 2021, 119, 743–761. [Google Scholar] [CrossRef]
- Rayfield, W.J.; Roush, D.J.; Chmielowski, R.A.; Tugcu, N.; Barakat, S.; Cheung, J.K. Prediction of viral filtration performance of monoclonal antibodies based on biophysical properties of feed. Biotechnol. Prog. 2015, 31, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Bolton, G.R.; Basha, J.; Lacasse, D.P. Achieving high mass-throughput of therapeutic proteins through parvovirus retentive filters. Biotechnol. Prog. 2010, 26, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Kosiol, P.; Kahrs, C.; Thom, V.; Ulbricht, M.; Hansmann, B. Investigation of virus retention by size exclusion membranes under different flow regimes. Biotechnol. Prog. 2019, 35, e2747. [Google Scholar] [CrossRef]
- Namila, F.N.U.; Zhang, D.; Traylor, S.; Nguyen, T.; Singh, N.; Wickramasinghe, R.; Qian, X. The effects of buffer condition on the fouling behavior of MVM virus filtration of an Fc-fusion protein. Biotechnol. Bioeng. 2019, 116, 2621–2631. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.; Tanenbaum, L.M.; Thirumangalathu, R.; Somani, S.; Zhang, K.; Kumar, V.; Amin, K.; Thakkar, S.V. Product-Specific Impact of Viscosity Modulating Formulation Excipients During Ultra-High Concentration Biotherapeutics Drug Product Development. J. Pharm. Sci. 2021, 110, 1077–1082. [Google Scholar] [CrossRef]
- Hu, Y.; Arora, J.; Joshi, S.B.; Esfandiary, R.; Middaugh, C.R.; Weis, D.D.; Volkin, D.B. Characterization of Excipient Effects on Reversible Self-Association, Backbone Flexibility, and Solution Properties of an IgG1 Monoclonal Antibody at High Concentrations: Part 1. J. Pharm. Sci. 2020, 109, 340–352. [Google Scholar] [CrossRef] [Green Version]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef]
- Philo, J.S.; Arakawa, T. Mechanisms of protein aggregation. Curr. Pharm. Biotechnol. 2009, 10, 348–351. [Google Scholar] [CrossRef]
- Chakroun, N.; Hilton, D.; Ahmad, S.S.; Platt, G.W.; Dalby, P.A. Mapping the Aggregation Kinetics of a Therapeutic Antibody Fragment. Mol. Pharm. 2016, 13, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Woll, A.K.; Hubbuch, J. Investigation of the reversibility of freeze/thaw stress-induced protein instability using heat cycling as a function of different cryoprotectants. Bioprocess Biosyst. Eng. 2020, 43, 1309–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorai, H.; Ganguly, S. Mammalian cell-produced therapeutic proteins: Heterogeneity derived from protein degradation. Curr. Opin. Biotechnol. 2014, 30, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Villanueva, J.F.; Diaz-Molina, R.; Garcia-Gonzalez, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [Green Version]
- Sule, S.V.; Cheung, J.K.; Antochshuk, V.; Bhalla, A.S.; Narasimhan, C.; Blaisdell, S.; Shameem, M.; Tessier, P.M. Solution pH that minimizes self-association of three monoclonal antibodies is strongly dependent on ionic strength. Mol. Pharm. 2012, 9, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.Y.; Janis, L.J. Influence of pH, buffer species, and storage temperature on physicochemical stability of a humanized monoclonal antibody LA298. Int. J. Pharm. 2006, 308, 46–51. [Google Scholar] [CrossRef]
- Salinas, B.A.; Sathish, H.A.; Shah, A.U.; Carpenter, J.F.; Randolph, T.W. Buffer-Dependent Fragmentation of a Humanized Full-Length Monoclonal Antibody. J. Pharm. Sci. 2010, 99, 2962–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, I.C.; Patel, A.R. Predicting the Agitation-Induced Aggregation of Monoclonal Antibodies Using Surface Tensiometry. Mol. Pharm. 2015, 12, 3184–3193. [Google Scholar] [CrossRef] [PubMed]
- Kueltzo, L.A.; Wang, W.; Randolph, T.W.; Carpenter, J.F. Effects of solution conditions, processing parameters, and container materials on aggregation of a monoclonal antibody during freeze-thawing. J. Pharm. Sci. 2008, 97, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Hawe, A.; Kasper, J.C.; Friess, W.; Jiskoot, W. Structural properties of monoclonal antibody aggregates induced by freeze–thawing and thermal stress. Eur. J. Pharm. Sci. 2009, 38, 79–87. [Google Scholar] [CrossRef]
- Barnard, J.G.; Kahn, D.; Cetlin, D.; Randolph, T.W.; Carpenter, J.F. Investigations into the fouling mechanism of parvovirus filters during filtration of freeze-thawed mAb drug substance solutions. J. Pharm. Sci. 2014, 103, 890–899. [Google Scholar] [CrossRef]
- Barnett, G.V.; Qi, W.; Amin, S.; Lewis, E.N.; Razinkov, V.I.; Kerwin, B.A.; Liu, Y.; Roberts, C.J. Structural Changes and Aggregation Mechanisms for Anti-Streptavidin IgG1 at Elevated Concentration. J. Phys. Chem. B 2015, 119, 15150–15163. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Prabakaran, P.; Chen, W.; Zhu, Z.; Feng, Y.; Dimitrov, D.S. Antibody Aggregation: Insights from Sequence and Structure. Antibodies 2016, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Protein aggregation and its inhibition in biopharmaceutics. Int. J. Pharm. 2005, 289, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Rey, M.; Lang, D.A. Aggregates in monoclonal antibody manufacturing processes. Biotechnol. Bioeng. 2011, 108, 1494–1508. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Havlíček, V. Protein Extraction and Precipitation. In Proteomic Profiling and Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; pp. 51–62. [Google Scholar] [CrossRef]
- Franco, R.; Daniela, G.; Fabrizio, M.; Ilaria, G.; Detlev, H. Influence of osmolarity and pH increase to achieve a reduction of monoclonal antibodies aggregates in a production process. Cytotechnology 1999, 29, 11–25. [Google Scholar] [CrossRef]
- Leckband, D.; Israelachvili, J. Intermolecular forces in biology. Q. Rev. Biophys. 2001, 34, 105–267. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.G.; Singh, S.; Randolph, T.W.; Carpenter, J.F. Subvisible particle counting provides a sensitive method of detecting and quantifying aggregation of monoclonal antibody caused by freeze-thawing: Insights into the roles of particles in the protein aggregation pathway. J. Pharm. Sci. 2011, 100, 492–503. [Google Scholar] [CrossRef]
- Bria, C.R.; Jones, J.; Charlesworth, A.; Ratanathanawongs Williams, S.K. Probing Submicron Aggregation Kinetics of an IgG Protein by Asymmetrical Flow Field-Flow Fractionation. J. Pharm. Sci. 2016, 105, 31–39. [Google Scholar] [CrossRef]
- Aboulaich, N.; Chung, W.K.; Thompson, J.H.; Larkin, C.; Robbins, D.; Zhu, M. A novel approach to monitor clearance of host cell proteins associated with monoclonal antibodies. Biotechnol. Prog. 2014, 30, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, A.H.; Moise, L.; De Groot, A.S. Of [Hamsters] and men: A new perspective on host cell proteins. Hum. Vaccin. Immunother. 2012, 8, 1172–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doneanu, C.E.; Xenopoulos, A.; Fadgen, K.; Murphy, J.; Skilton, S.J.; Prentice, H.; Stapels, M.; Chen, W. Analysis of host-cell proteins in biotherapeutic proteins by comprehensive online two-dimensional liquid chromatography/mass spectrometry. MAbs 2012, 4, 24–44. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.A.; Hinckley, P. Host cell protein clearance during protein a chromatography: Development of an improved column wash step. Biotechnol. Prog. 2008, 24, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Nogal, B.; Chhiba, K.; Emery, J.C. Select host cell proteins coelute with monoclonal antibodies in protein A chromatography. Biotechnol. Prog. 2012, 28, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, D.; Dhillon, H.; Slaney, T.; Song, H.; Boux, H.; Mehta, S.; Zhang, L.; Valdez, A.; Krishnamurthy, G. Identification of a host cell protein impurity in therapeutic protein, P1. J. Pharm. Biomed. Anal. 2017, 141, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Goetze, A.M.; Cui, H.; Wylie, J.; Trimble, S.; Hewig, A.; Flynn, G.C. Comprehensive tracking of host cell proteins during monoclonal antibody purifications using mass spectrometry. MAbs 2014, 6, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.X.; Zhang, Y.; Stansberry-Perkins, K.; Buko, A.; Bai, S.; Nguyen, V.; Brader, M.L. Fragmentation of a highly purified monoclonal antibody attributed to residual CHO cell protease activity. Biotechnol. Bioeng. 2011, 108, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Charcosset, C. 4—Virus filtration. In Membrane Processes in Biotechnology and Pharmaceutics; Charcosset, C., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 143–167. [Google Scholar] [CrossRef]
- Gilgunn, S.; El-Sabbahy, H.; Albrecht, S.; Gaikwad, M.; Corrigan, K.; Deakin, L.; Jellum, G.; Bones, J. Identification and tracking of problematic host cell proteins removed by a synthetic, highly functionalized nonwoven media in downstream bioprocessing of monoclonal antibodies. J. Chromatogr. A 2019, 1595, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Kornecki, M.; Mestmacker, F.; Zobel-Roos, S.; Heikaus de Figueiredo, L.; Schluter, H.; Strube, J. Host Cell Proteins in Biologics Manufacturing: The Good, the Bad, and the Ugly. Antibodies 2017, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luhrs, K.A.; Harris, D.A.; Summers, S.; Parseghian, M.H. Evicting hitchhiker antigens from purified antibodies. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 1543–1552. [Google Scholar] [CrossRef]
- Chen, R.H.; Huang, C.J.; Newton, B.S.; Ritter, G.; Old, L.J.; Batt, C.A. Factors affecting endotoxin removal from recombinant therapeutic proteins by anion exchange chromatography. Protein Expr. Purif. 2009, 64, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, L.; Cheng, L.-H.; Xu, K.; Xu, Q.-P.; Chen, H.-L.; Lai, J.-Y.; Tung, K.-L. Extracorporeal endotoxin removal by novel l-serine grafted PVDF membrane modules. J. Membr. Sci. 2012, 405–406, 104–112. [Google Scholar] [CrossRef]
- Ongkudon, C.M.; Chew, J.H.; Liu, B.; Danquah, M.K. Chromatographic Removal of Endotoxins: A Bioprocess Engineer’s Perspective. ISRN Chromatogr. 2012, 2012, 649746. [Google Scholar] [CrossRef] [Green Version]
- Petsch, D.; Anspach, F.B. Endotoxin removal from protein solutions. J. Biotechnol. 2000, 76, 97–119. [Google Scholar] [CrossRef]
- Ritzén, U.; Rotticci-Mulder, J.; Strömberg, P.; Schmidt, S.R. Endotoxin reduction in monoclonal antibody preparations using arginine. J. Chromatogr. B 2007, 856, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Kahle, J.; Watzig, H. Determination of protein charge variants with (imaged) capillary isoelectric focusing and capillary zone electrophoresis. Electrophoresis 2018, 39, 2492–2511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Park, S.-H.; Boucher, S.; Higgins, E.; Lee, K.; Edmunds, T. N-linked oligosaccharide analysis of glycoprotein bands from isoelectric focusing gels. Anal. Biochem. 2004, 335, 10–16. [Google Scholar] [CrossRef]
- Wingfield, P.T. Overview of the purification of recombinant proteins. Curr. Protoc. Protein Sci. 2015, 80, 6.1.1–6.1.35. [Google Scholar] [CrossRef] [Green Version]
- Vlasak, J.; Ionescu, R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr. Pharm. Biotechnol. 2008, 9, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Kahle, J.; Zagst, H.; Wiesner, R.; Watzig, H. Comparative charge-based separation study with various capillary electrophoresis (CE) modes and cation exchange chromatography (CEX) for the analysis of monoclonal antibodies. J. Pharm. Biomed. Anal. 2019, 174, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Lamp, J.; Xia, Q.; Zhang, Y. Capillary Isoelectric Focusing-Mass Spectrometry Method for the Separation and Online Characterization of Intact Monoclonal Antibody Charge Variants. Anal. Chem. 2018, 90, 2246–2254. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ren, W.; Zong, L.; Zhang, J.; Wang, Y. Characterization of recombinant monoclonal antibody charge variants using WCX chromatography, icIEF and LC-MS/MS. Anal. Biochem. 2019, 564–565, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.M.; Berger, L.; Nerkamp, J.; Scheler, S.; Nehring, S.; Friess, W. Identification of Monoclonal Antibody Variants Involved in Aggregate Formation—Part 1: Charge Variants. Eur. J. Pharm. Biopharm. 2020, 158, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Rohani, M.M.; Zydney, A.L. Role of electrostatic interactions during protein ultrafiltration. Adv. Colloid. Interface Sci. 2010, 160, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Roush, D.; Cramer, S.M. The effect of pH on antibody retention in multimodal cation exchange chromatographic systems. J. Chromatogr. A 2020, 1617, 460838. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.R.; Kirkland, J.J.; Dolan, J.W. Introduction to Modern Liquid Chromatography, 3rd ed.; Wiley-VCH: Hoboken, NJ, USA, 2010; pp. 584–618. [Google Scholar]
- Lehermayr, C.; Mahler, H.C.; Mader, K.; Fischer, S. Assessment of net charge and protein-protein interactions of different monoclonal antibodies. J. Pharm. Sci. 2011, 100, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Fekete, S.; Veuthey, J.L.; Beck, A.; Guillarme, D. Hydrophobic interaction chromatography for the characterization of monoclonal antibodies and related products. J. Pharm. Biomed. Anal. 2016, 130, 3–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, A.; Olszowy, P.; Ciborowski, P. Biomolecules. In Proteomic Profiling and Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; pp. 7–24. [Google Scholar] [CrossRef]
- Fekete, S.; Guillarme, D.; Sandra, P.; Sandra, K. Chromatographic, Electrophoretic, and Mass Spectrometric Methods for the Analytical Characterization of Protein Biopharmaceuticals. Anal. Chem. 2016, 88, 480–507. [Google Scholar] [CrossRef] [PubMed]
- Borisov, O.V.; Alvarez, M.; Carroll, J.A.; Brown, P.W. Sequence Variants and Sequence Variant Analysis in Biotherapeutic Proteins. In State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2. Biopharmaceutical Characterization: The NISTmAb Case Study; American Chemical Society: Washington, DC, USA, 2015; Volume 1201, pp. 63–117. [Google Scholar]
- Hinkle, J.D.; D’Ippolito, R.A.; Panepinto, M.C.; Wang, W.H.; Bai, D.L.; Shabanowitz, J.; Hunt, D.F. Unambiguous Sequence Characterization of a Monoclonal Antibody in a Single Analysis Using a Nonspecific Immobilized Enzyme Reactor. Anal. Chem. 2019, 91, 13547–13554. [Google Scholar] [CrossRef]
- Flynn, G.C.; Chen, X.; Liu, Y.D.; Shah, B.; Zhang, Z. Naturally occurring glycan forms of human immunoglobulins G1 and G2. Mol. Immunol. 2010, 47, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Srebalus Barnes, C.A.; Lim, A. Applications of mass spectrometry for the structural characterization of recombinant protein pharmaceuticals. Mass Spectrom. Rev. 2007, 26, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Herschel, T.; El-Armouche, A.; Weber, S. Monoclonal antibodies, overview and outlook of a promising therapeutic option. Dtsch. Med. Wochenschr. 2016, 141, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D. Edelman’s view on the discovery of antibodies. Immunol. Lett. 2015, 164, 72–75. [Google Scholar] [CrossRef]
- Dicker, M.; Strasser, R. Using glyco-engineering to produce therapeutic proteins. Expert Opin. Biol. 2015, 15, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Zydney, A.L. Perspectives on integrated continuous bioprocessing—opportunities and challenges. Curr. Opin. Chem. Eng. 2015, 10, 8–13. [Google Scholar] [CrossRef]
- Rowe, L.; El Khoury, G.; Lowe, C.R. Affinity Chromatography: Historical and Prospective Overview. Biopharm. Prod. Technol. 2012, 1, 223–282. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Liu, Y.H.; Richardson, D.; Li, H.; Shameem, M.; Yang, X. Simultaneous monitoring of oxidation, deamidation, isomerization, and glycosylation of monoclonal antibodies by liquid chromatography-mass spectrometry method with ultrafast tryptic digestion. MAbs 2016, 8, 1477–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Kim, S.M.; Ruzanski, R.; Chen, Y.; Moses, S.; Ling, W.L.; Li, X.; Wang, S.C.; Li, H.; Ambrogelly, A.; et al. Ultrafast and high-throughput N-glycan analysis for monoclonal antibodies. MAbs 2016, 8, 706–717. [Google Scholar] [CrossRef] [Green Version]
- Strohl, W.R.; Strohl, L.M. Therapeutic Antibody Engineering: Current and Future Advances Driving the Strongest Growth Area in the Pharmaceutical Industry; Elsevier Science & Technology: Cambridge, UK, 2012; Volume 11. [Google Scholar]
- Radhakrishnan, D.; Robinson, A.S.; Ogunnaike, B.A. Controlling the Glycosylation Profile in mAbs Using Time-Dependent Media Supplementation. Antibodies 2017, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Ivarsson, M.; Villiger, T.K.; Morbidelli, M.; Soos, M. Evaluating the impact of cell culture process parameters on monoclonal antibody N-glycosylation. J. Biotechnol. 2014, 188, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Song, H.; Slaney, T.; Ludwig, R.; Tao, L.; Das, T. Characterization of Protein Therapeutics by Mass Spectrometry. In Protein Analysis Using Mass Spectrometry: Accelerating Protein Biotherapeutics from Lab to Patient; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 221–249. [Google Scholar] [CrossRef]
- Gagneux, P.; Varki, A. Evolutionary considerations in relating oligosaccharide diversity to biological function. Glycobiology 1999, 9, 747–755. [Google Scholar] [CrossRef]
- Barinka, C.; Sacha, P.; Sklenar, J.; Man, P.; Bezouska, K.; Slusher, B.S.; Konvalinka, J. Identification of the N-glycosylation sites on glutamate carboxypeptidase II necessary for proteolytic activity. Protein Sci. 2004, 13, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrázek, H.; Weignerová, L.; Bojarová, P.; Novák, P.; Vaněk, O.; Bezouska, K. Carbohydrate synthesis and biosynthesis technologies for cracking of the glycan code: Recent advances. Biotechnol. Adv. 2012, 31, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Qiu, H. The Mechanistic Impact of N-Glycosylation on Stability, Pharmacokinetics, and Immunogenicity of Therapeutic Proteins. J. Pharm. Sci. 2019, 108, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Wada, R.; Matsui, M.; Kawasaki, N. Influence of N-glycosylation on effector functions and thermal stability of glycoengineered IgG1 monoclonal antibody with homogeneous glycoforms. MAbs 2019, 11, 350–372. [Google Scholar] [CrossRef]
- Planinc, A.; Bones, J.; Dejaegher, B.; Van Antwerpen, P.; Delporte, C. Glycan characterization of biopharmaceuticals: Updates and perspectives. Anal. Chim. Acta 2016, 921, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Sola, R.J.; Griebenow, K. Effects of glycosylation on the stability of protein pharmaceuticals. J. Pharm. Sci. 2009, 98, 1223–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhu, J.; Lu, H. Antibody glycosylation: Impact on antibody drug characteristics and quality control. Appl. Microbiol. Biotechnol. 2020, 104, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gaza-Bulseco, G.; Faldu, D.; Chumsae, C.; Sun, J. Heterogeneity of Monoclonal Antibodies. J. Pharm. Sci. 2008, 97, 2426–2447. [Google Scholar] [CrossRef] [PubMed]
- Hari, S.B.; Lau, H.; Razinkov, V.I.; Chen, S.; Latypov, R.F. Acid-induced aggregation of human monoclonal IgG1 and IgG2: Molecular mechanism and the effect of solution composition. Biochemistry 2010, 49, 9328–9338. [Google Scholar] [CrossRef] [PubMed]
- Alsenaidy, M.A.; Okbazghi, S.Z.; Kim, J.H.; Joshi, S.B.; Middaugh, C.R.; Tolbert, T.J.; Volkin, D.B. Physical stability comparisons of IgG1-Fc variants: Effects of N-glycosylation site occupancy and Asp/Gln residues at site Asn 297. J. Pharm. Sci. 2014, 103, 1613–1627. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Bechtel, C.; Bill, J.; Liu, H.; Liu, J.; McDonald, D.; Pai, S.; Radhamohan, A.; Renslow, R.; Thayer, B.; et al. Increasing parvovirus filter throughput of monoclonal antibodies using ion exchange membrane adsorptive pre-filtration. Biotechnol. Bioeng. 2010, 106, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Yigzaw, Y.; Hinckley, P.; Hewig, A.; Vedantham, G. Ion exchange chromatography of proteins and clearance of aggregates. Curr. Pharm. Biotechnol. 2009, 10, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Saraswat, M.; Musante, L.; Ravida, A.; Shortt, B.; Byrne, B.; Holthofer, H. Preparative purification of recombinant proteins: Current status and future trends. Biomed. Res. Int. 2013, 2013, 312709. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Tsumoto, K.; Nagase, K.; Ejima, D. The effects of arginine on protein binding and elution in hydrophobic interaction and ion-exchange chromatography. Protein Expr. Purif. 2007, 54, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Jabra, M.G.; Tao, Y.; Moomaw, J.F.; Yu, Z.; Hotovec, B.J.; Geng, S.B.; Zydney, A.L. pH and excipient profiles during formulation of highly concentrated biotherapeutics using bufferless media. Biotechnol. Bioeng. 2020, 117, 3390–3399. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug Classification | Examples | First Approval by FDA | Manufacturer |
|---|---|---|---|
| Monoclonal antibodies | Pembrolizumab | 2014 | Merck |
| Nivolumab | 2014 | Bristol Myers Squibb | |
| Aducanumab | 2021 | Biogen | |
| Avelumab | 2017 | EMD Serono | |
| Omalizumab | 2003 | Genentech | |
| Adalimumab | 2002 | Abbvie | |
| Tezepelumab-ekko | 2021 | Amgen/AstraZeneca | |
| Fc-fusion proteins | Abatacept | 2021 | Bristol Myers Squibb |
| Aflibercept | 2011 | Regeneron | |
| Alefacept | 2003 | Biogen | |
| Etanercept | 1998 | Amgen | |
| Rilonacept | 2008 | Regeneron | |
| Cytokines | Darbepoetin alfa | 2011 | Amgen |
| Interferon beta-1a | 2003 | Biogen | |
| Epoetin alfa | 2011 | Amgen | |
| Enzymes | Agalsidase beta | 2003 | Genzyme |
| Human DNase | 1993 | Genentech | |
| Laronidase | 2003 | Biomarin | |
| Tenecteplase | 2000 | Genentech | |
| Hormones | Choriogonadotropin alfa | 2000 | EMD Serono |
| Follitropin alfa | 2004 | EMD Serono | |
| Osteogenic protein-1 | 2001 | Stryker Biotech | |
| Thyrotropin alfa | 1998 | Genzyme |
| Name of Virus | Diameter (nm) |
|---|---|
| Animal parvoviruses (non-enveloped DNA viruses, bovine, canine, or porcine) | 18–24 |
| Poliovirus (picornavirus, non-enveloped RNA virus) | 25–30 |
| Encephalomyocarditis virus (EMC, picornavirus, non-enveloped RNA virus) | 25–30 |
| Feline calicivirus (calicivirus, non-enveloped RNA virus) | 35–39 |
| Bovine viral diarrhea virus (BVDV, flavivirus, enveloped RNA virus) | 40–60 |
| SV40 (simian vacuolating virus 40, polyomavirus, non-enveloped DNA virus) | 45–55 |
| Sindbis virus (togavirus, enveloped RNA virus) | 60–70 |
| Reovirus (non-enveloped RNA virus) | 60–80 |
| Herpes simplex virus (HSV, Herpesviridae, enveloped DNA virus) | 150 |
| Pseudorabies virus (PRV, Herpesviridae, enveloped DNA virus) | 120–200 |
| Filter | Manufacturer | Membrane Material | Configuration | Comments |
|---|---|---|---|---|
| Planova 15 N, 20 N | Asahi Kasei Bioprocess | Regenerated cellulose | Asymmetric single-layer hollow fibers | Parvovirus filter |
| Planova 35 N | Asahi Kasei Bioprocess | Regenerated cellulose | Asymmetric single-layer hollow fibers | Retrovirus filter |
| Planova BioEX | Asahi Kasei Bioprocess | Hydrophilized PVDF | Asymmetric single-layer hollow fibers | Parvovirus filter |
| Viresolve NFR | MilliporeSigma | Polyethersulfone | Asymmetric triple-layer pleated sheets | Retrovirus filter |
| Viresolve Pro | MilliporeSigma | Polyethersulfone | Asymmetric double-layer flat sheets | Parvovirus filter |
| Pegasus SV4 | Pall Corporation | Hydrophilized PVDF | Symmetric double-layer pleated sheets | Parvovirus filter |
| Pegasus Prime | Pall Corporation | Polyethersulfone | Pleated sheets | Parvovirus filter |
| Ultipor VF DV20 | Pall Corporation | Hydrophilized PVDF | Symmetric double-layer pleated sheets | Parvovirus filter |
| Ultipor VF DV50 | Pall Corporation | Hydrophilized PVDF | Symmetric double-layer pleated sheets | Retrovirus filter |
| Virosart HC | Sartorius AG | Polyethersulfone | Asymmetric double-layer pleated sheets | Parvovirus filter |
| Virosart HF | Sartorius AG | Modified polyethersulfone | Asymmetric single-layer hollow fibers | Parvovirus filter |
| Prefilter | Material | Mechanism of Action | Manufacturer |
|---|---|---|---|
| Planova 75 N | Regenerated cellulose | Size exclusion, removal of small aggregates | Asahi Kasei Bioprocess |
| Bottle top 0.1/0.22 µm | Polyethersulfone | Size exclusion, removal of large aggregates | Multiple |
| Pegasus Protect | Nylon | Size exclusion, removal of large aggregates | Pall |
| Sartobind Q | Quaternary ammonium ligands | Anion exchange | Sartorius AG |
| Sartobind S | Sulfonic acid ligands | Cation exchange | Sartorius AG |
| Sartobind phenyl | Phenyl ligands | Hydrophobic interaction | Sartorius AG |
| Viresolve Pro Shield | Surface modified PES | Size exclusion, ion exchange (cation) | MilliporeSigma |
| Viresolve Pro Shield H | Surface modified PES | Size exclusion, hydrophobic interaction | MilliporeSigma |
| Viresolve Prefilter | Diatomaceous earth, cellulose fibers, and a cationic imine binder | Cation exchange, size exclusion, hydrophobic interaction, ion exchange | MilliporeSigma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isu, S.; Qian, X.; Zydney, A.L.; Wickramasinghe, S.R. Process- and Product-Related Foulants in Virus Filtration. Bioengineering 2022, 9, 155. https://doi.org/10.3390/bioengineering9040155
Isu S, Qian X, Zydney AL, Wickramasinghe SR. Process- and Product-Related Foulants in Virus Filtration. Bioengineering. 2022; 9(4):155. https://doi.org/10.3390/bioengineering9040155
Chicago/Turabian StyleIsu, Solomon, Xianghong Qian, Andrew L. Zydney, and S. Ranil Wickramasinghe. 2022. "Process- and Product-Related Foulants in Virus Filtration" Bioengineering 9, no. 4: 155. https://doi.org/10.3390/bioengineering9040155