

A Multi-Specific DARPin Potently Neutralizes Shiga Toxin 2 via Simultaneous Modulation of Both Toxin Subunits

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. DARPin Library Construction
2.3. Phage Panning
2.4. Functional Screening
2.5. Protein Purification
2.6. Vero E6 Toxin Challenging Assay
2.7. In Vitro Catalytic Activity Assay
2.8. ELISA
2.9. Affinity Measurement
2.10. Cryo-EM Sample Preparation
2.11. Cryo-EM Data Collection
2.12. Image Processing
2.13. Model Building
2.14. Data Availability
2.15. Stx Toxin Challenge in Mice
3. Results
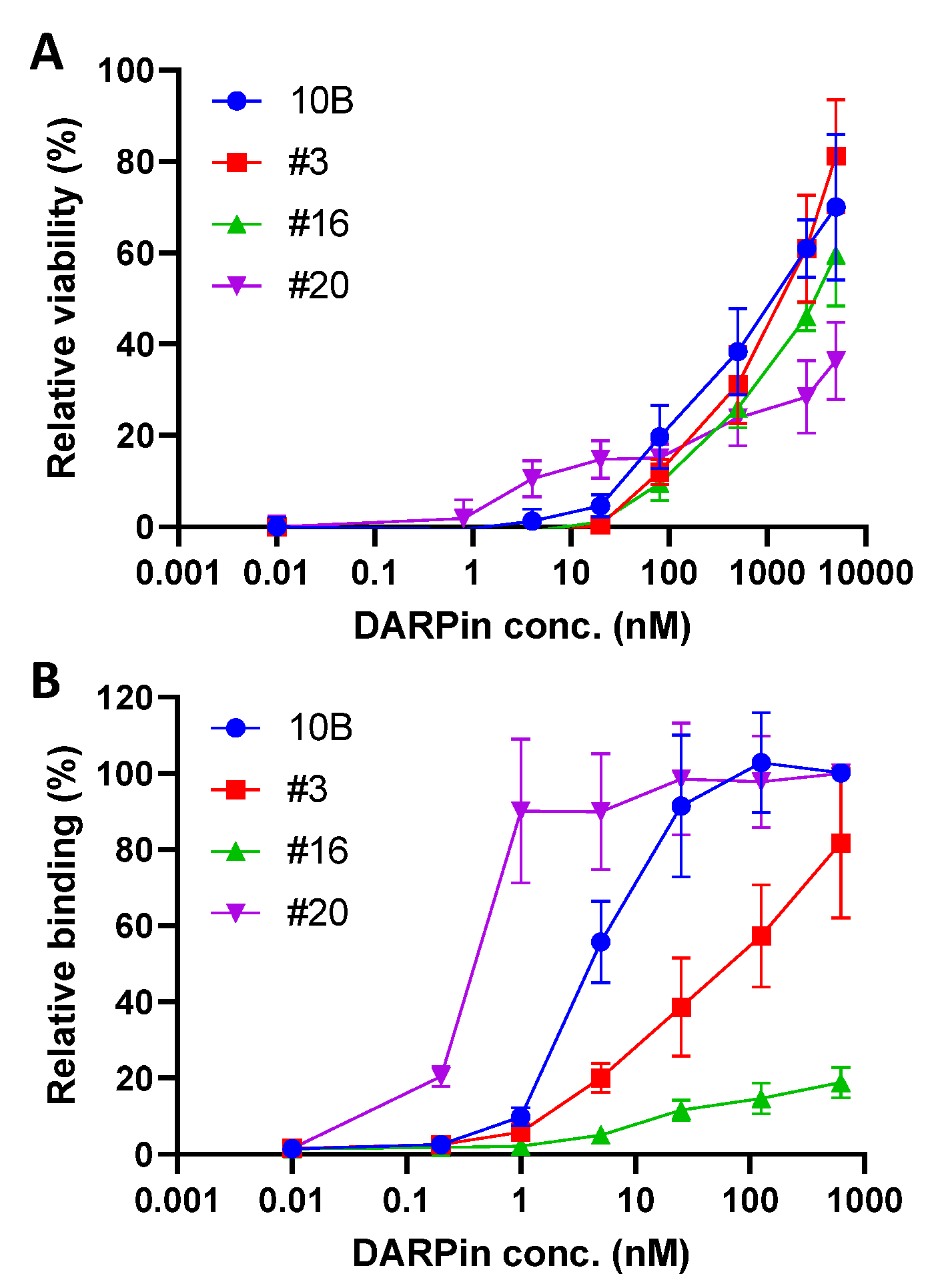
3.1. Selection of Monomeric Stx2a-Neutralizing DARPins
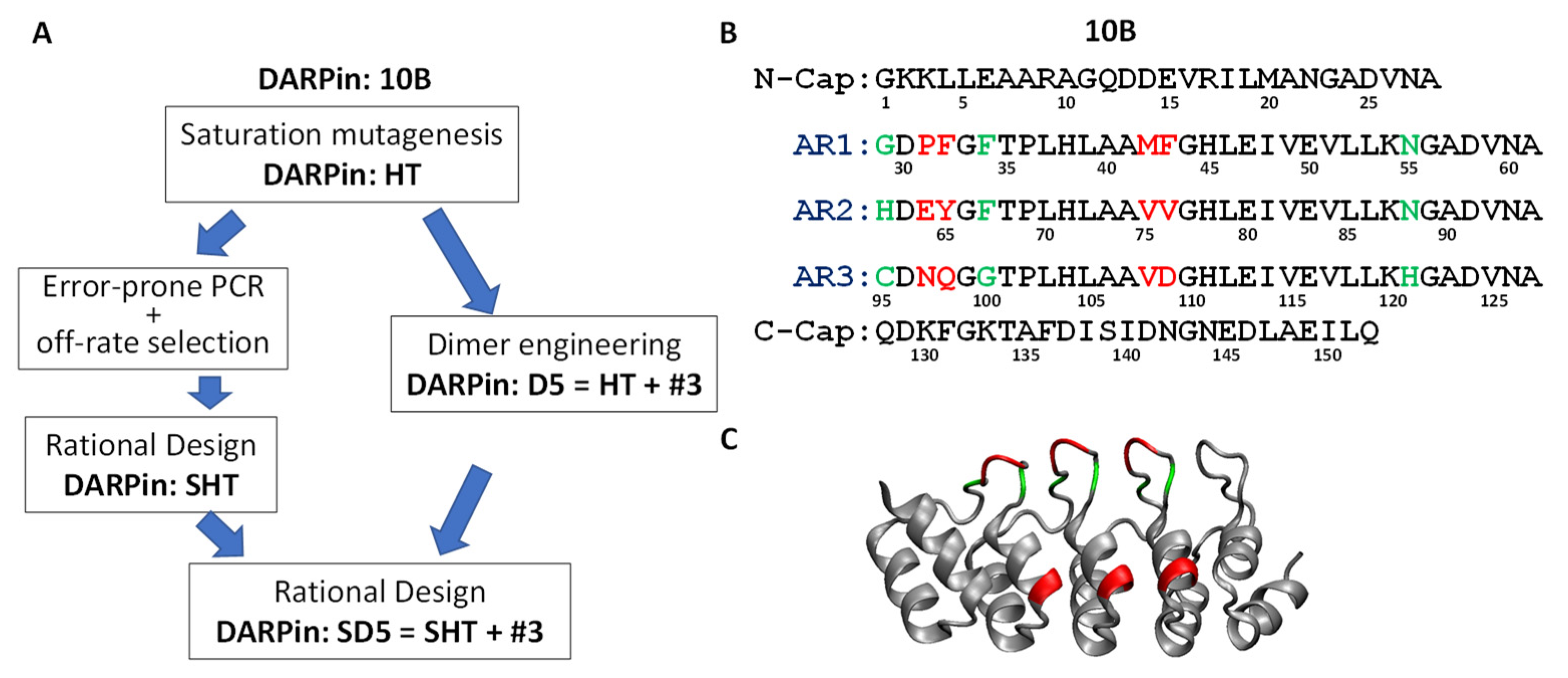
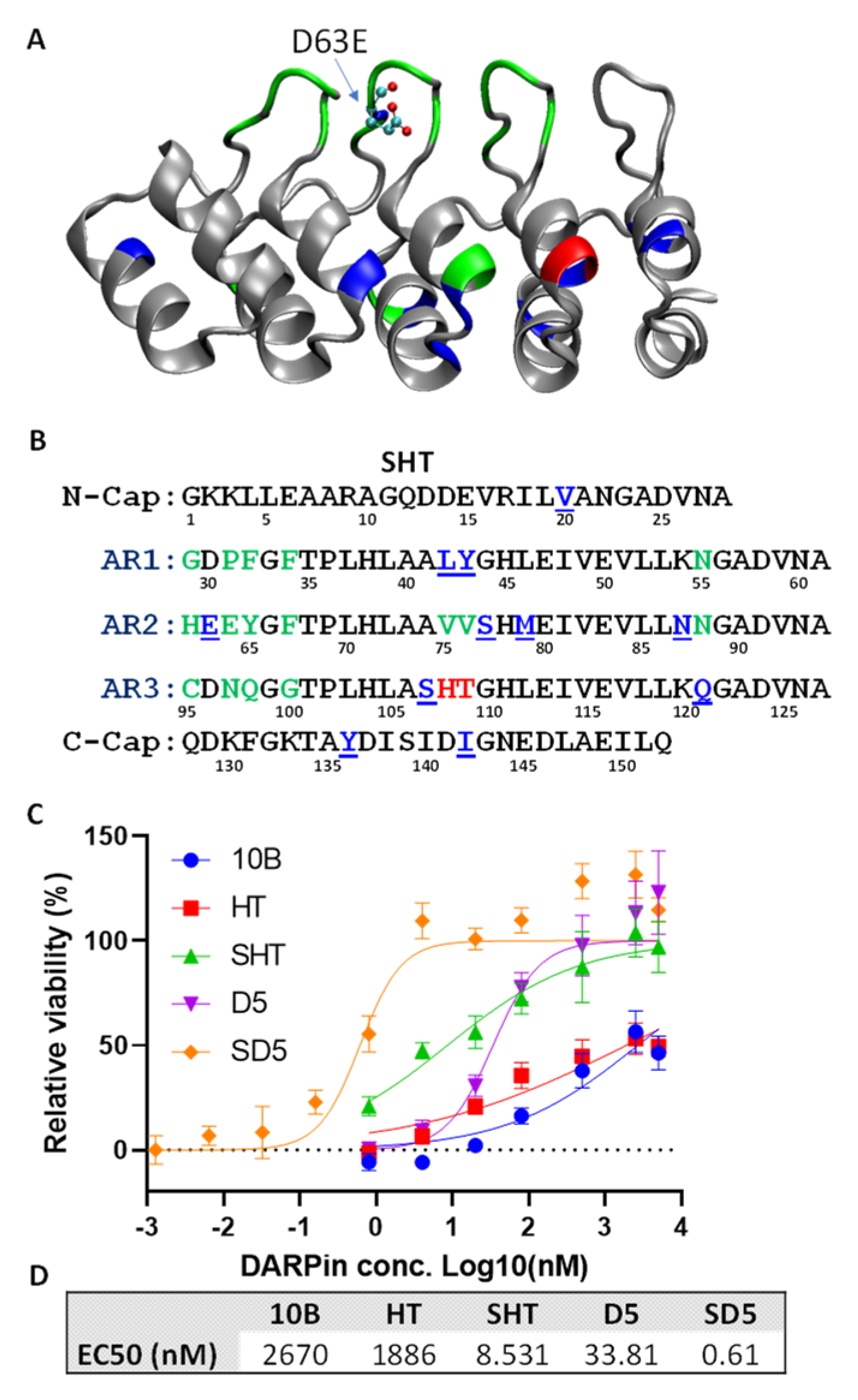
3.2. Systematic Affinity Maturation
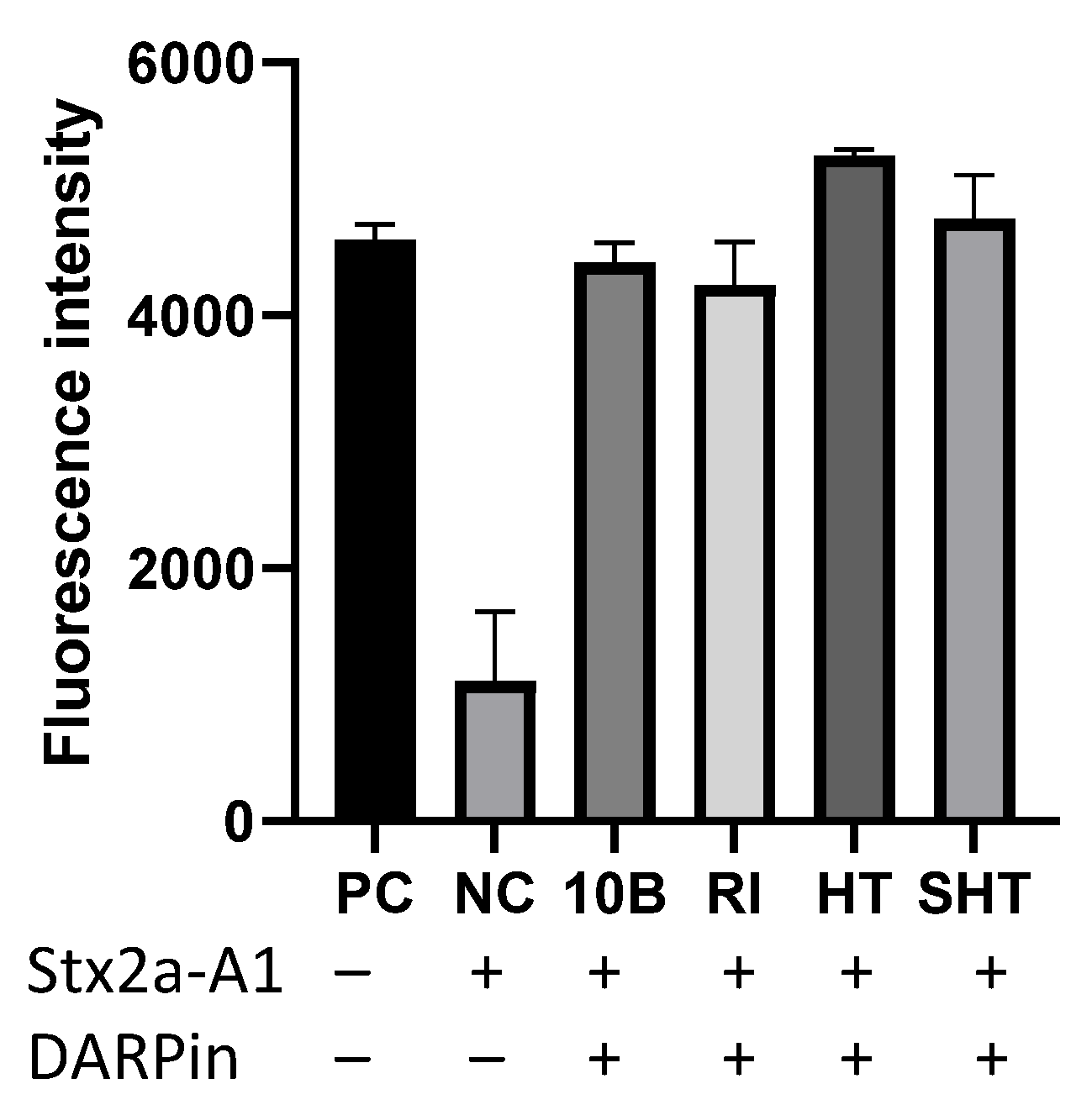
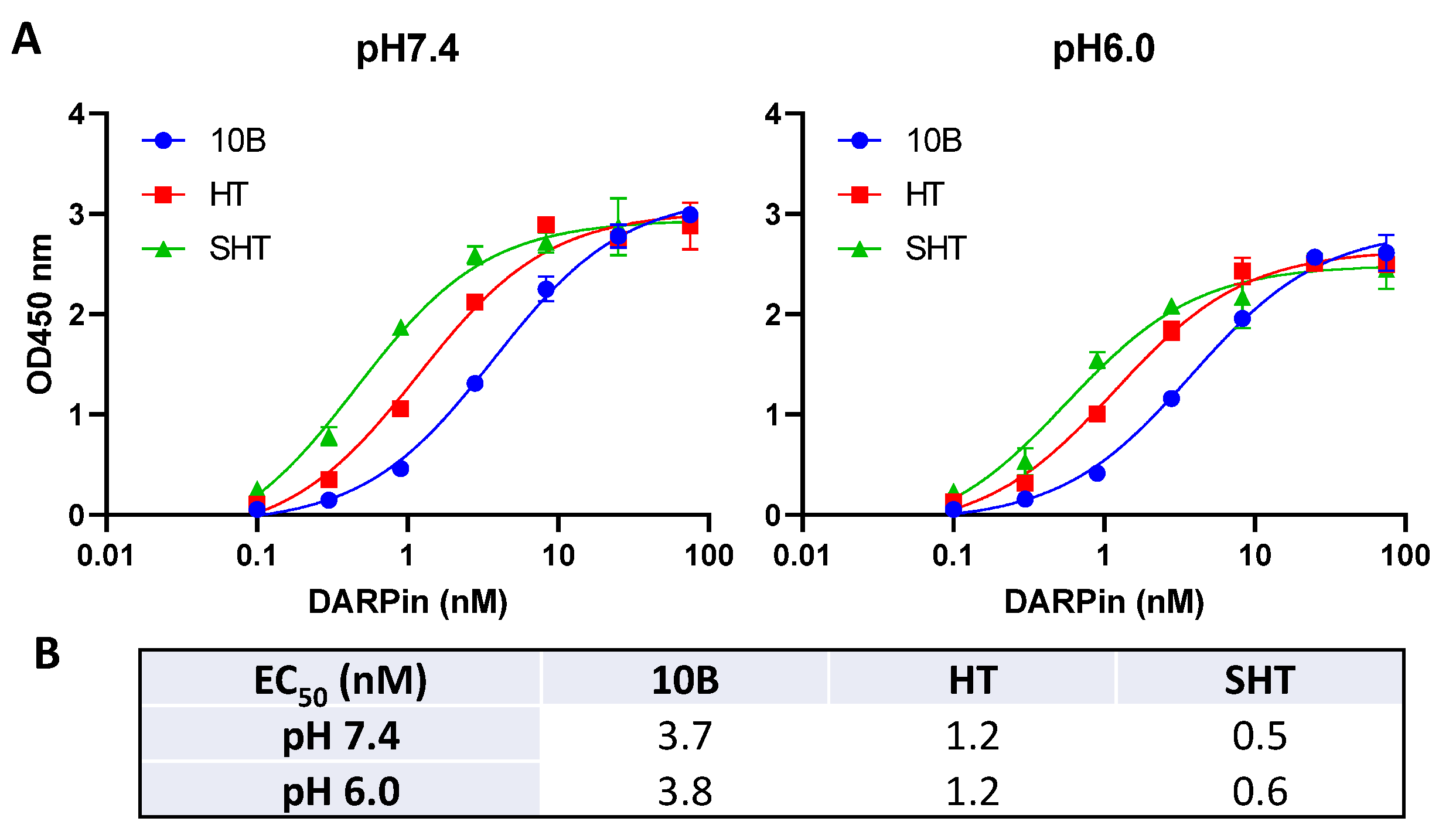
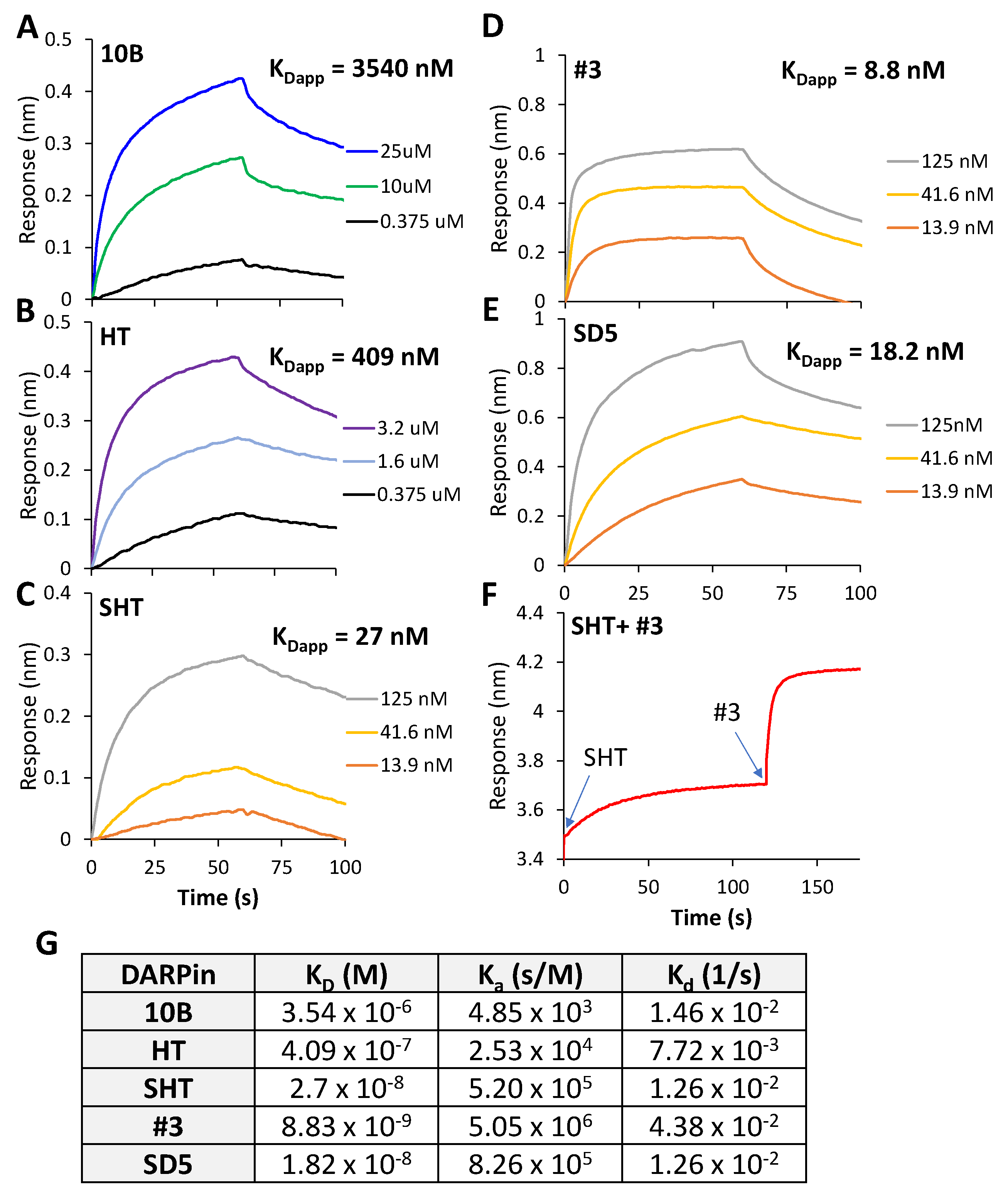
3.3. In Vitro Characterization of DARPin Molecules
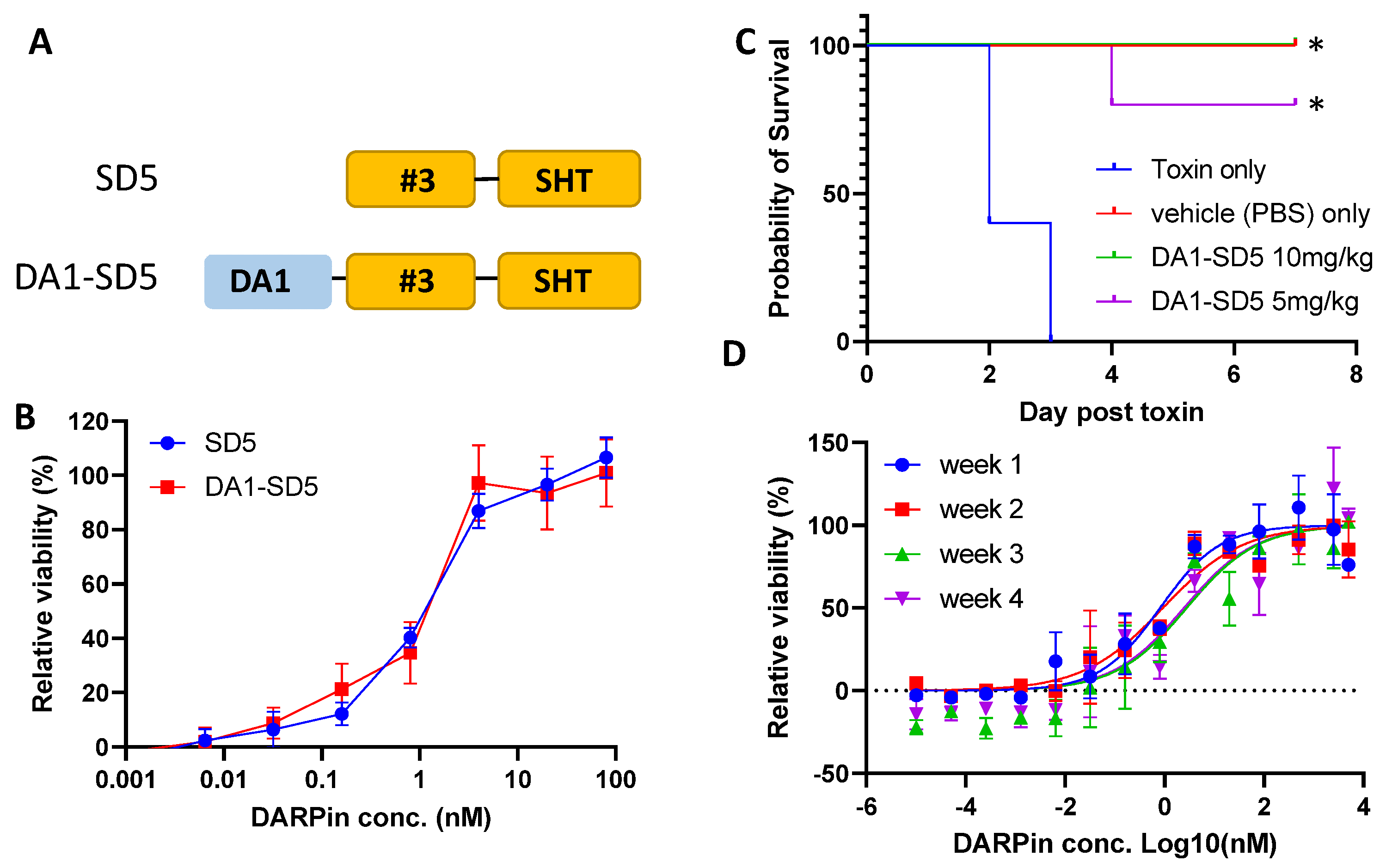
3.4. In Vivo Efficacy of DARPin
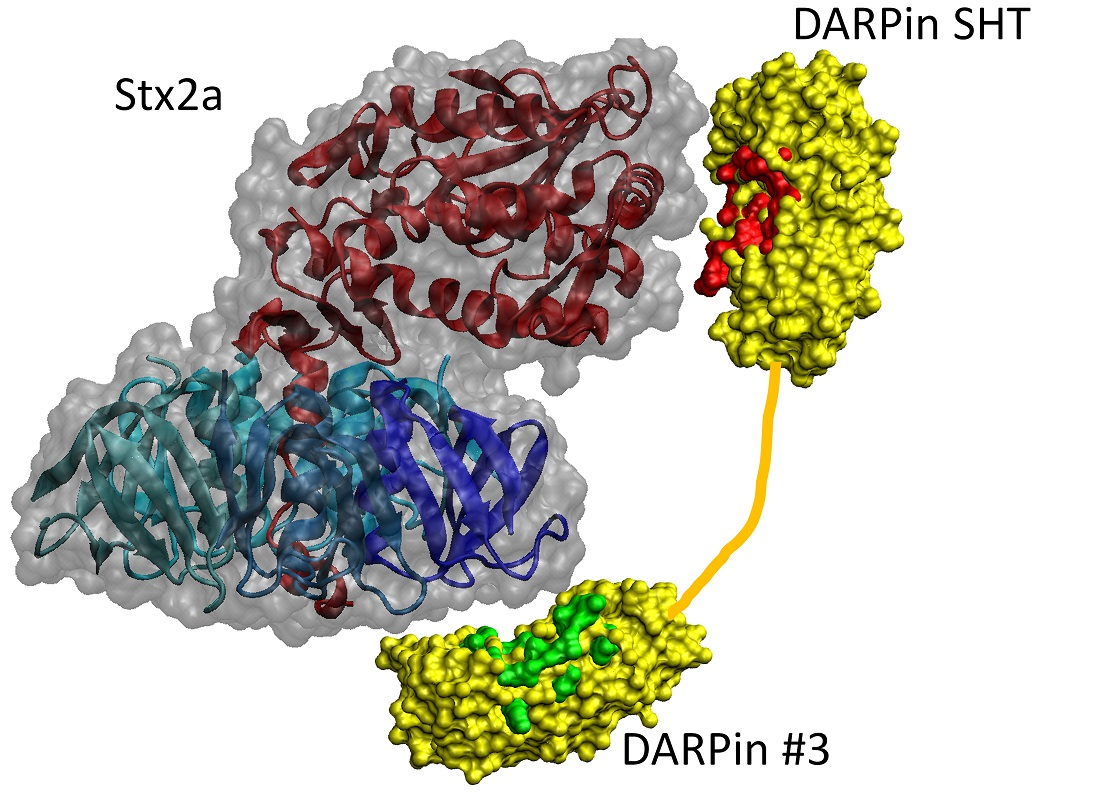
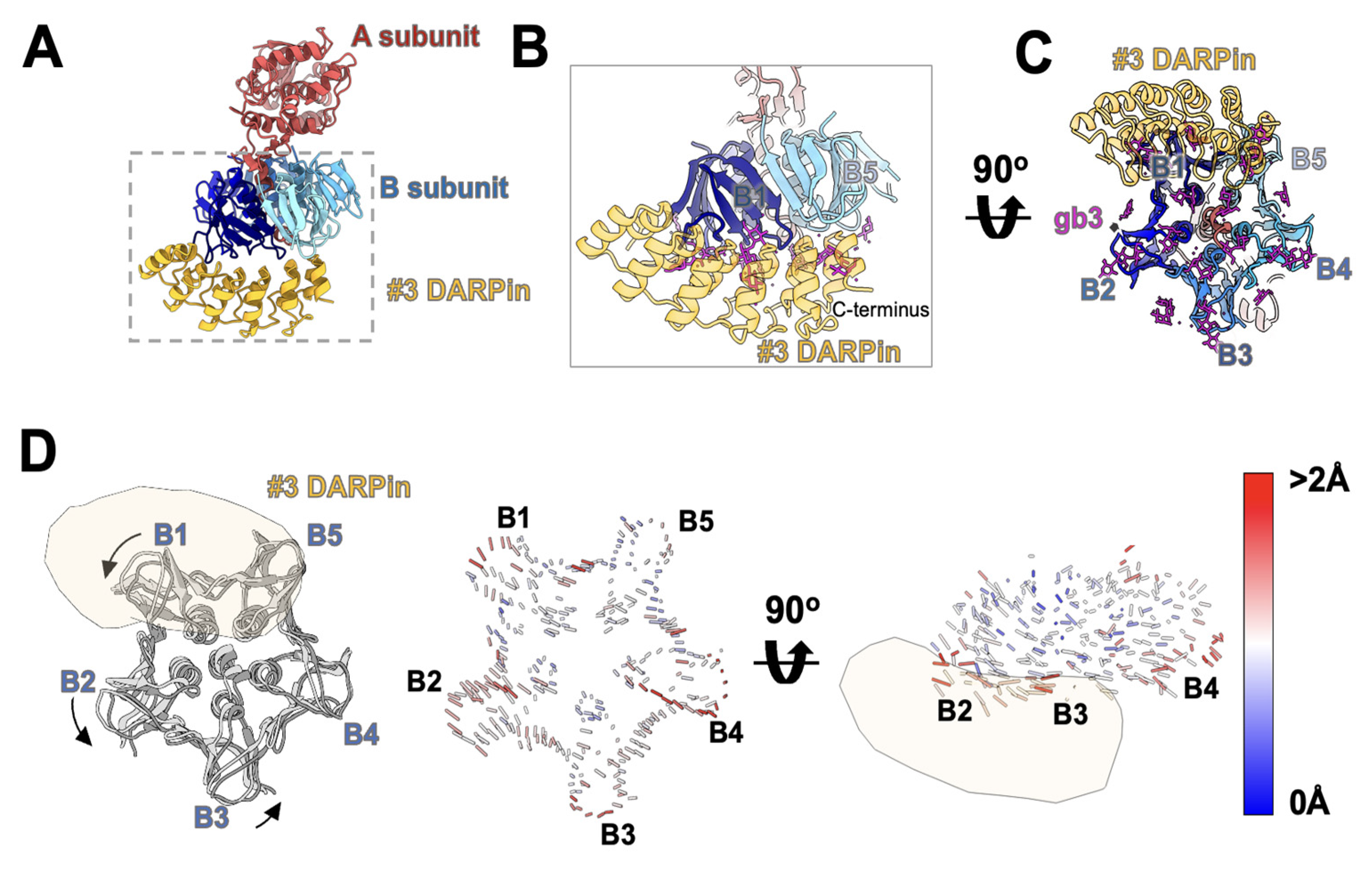
3.5. Cryo-EM Study of DARPin #3 and Stx2a
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Center for Disease Control and Prevention. E. coli Q&A. Available online: https://www.cdc.gov/ecoli/general/index.html (accessed on 22 August 2022).
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Melton-Celsa, A.; Mohawk, K.; Teel, L.; O’Brien, A. Pathogenesis of Shiga-toxin producing Escherichia coli. Curr. Top Microbiol. Immunol. 2012, 357, 67–103. [Google Scholar] [CrossRef]
- Trofa, A.F.; Ueno-Olsen, H.; Oiwa, R.; Yoshikawa, M. Dr. Kiyoshi Shiga: Discoverer of the dysentery bacillus. Clin. Infect. Dis. 1999, 29, 1303–1306. [Google Scholar] [CrossRef]
- Dubos, R.J.; Geiger, J.W. Preparation and properties of Shiga toxin and toxoid. J. Exp. Med. 1946, 84, 143–156. [Google Scholar] [CrossRef]
- McPherson, M.; Kirk, M.D.; Raupach, J.; Combs, B.; Butler, J.R. Economic costs of Shiga toxin-producing Escherichia coli infection in Australia. Foodborne Pathog. Dis. 2011, 8, 55–62. [Google Scholar] [CrossRef]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef]
- Tesh, V.L. Induction of apoptosis by Shiga toxins. Future Microbiol. 2010, 5, 431–453. [Google Scholar] [CrossRef]
- Dundas, S.; Todd, W.T.; Neill, M.A.; Tarr, P.I. Using antibiotics in suspected haemolytic-uraemic syndrome: Antibiotics should not be used in Escherichia coli O157:H7 infection. BMJ 2005, 330, 1209. [Google Scholar] [CrossRef]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef]
- Zhang, X.; McDaniel, A.D.; Wolf, L.E.; Keusch, G.T.; Waldor, M.K.; Acheson, D.W. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J. Infect. Dis. 2000, 181, 664–670. [Google Scholar] [CrossRef]
- Wong, C.S.; Jelacic, S.; Habeeb, R.L.; Watkins, S.L.; Tarr, P.I. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N. Engl. J. Med. 2000, 342, 1930–1936. [Google Scholar] [CrossRef]
- Ahn, C.K.; Holt, N.J.; Tarr, P.I. Shiga-toxin producing Escherichia coli and the hemolytic uremic syndrome: What have we learned in the past 25 years? Adv. Exp. Med. Biol. 2009, 634, 1–17. [Google Scholar]
- Center for Disease Control. Available online: http://www.cdc.gov/ecoli/ (accessed on 22 August 2022).
- Scheutz, F.; Teel, L.D.; Beutin, L.; Pierard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef]
- Yang, X.; Bai, X.; Zhang, J.; Sun, H.; Fu, S.; Fan, R.; He, X.; Scheutz, F.; Matussek, A.; Xiong, Y. Escherichia coli strains producing a novel Shiga toxin 2 subtype circulate in China. Int. J. Med. Microbiol. 2020, 310, 151377. [Google Scholar] [CrossRef]
- Fuller, C.A.; Pellino, C.A.; Flagler, M.J.; Strasser, J.E.; Weiss, A.A. Shiga Toxin Subtypes Display Dramatic Differences in Potency. Infect. Immun. 2011, 79, 1329–1337. [Google Scholar] [CrossRef]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [CrossRef]
- Kawano, K.; Okada, M.; Haga, T.; Maeda, K.; Goto, Y. Relationship between pathogenicity for humans and stx genotype in Shiga toxin-producing Escherichia coli serotype O157. Eur. J. Clin. Microbiol. Infect. Dis. 2008, 27, 227–232. [Google Scholar] [CrossRef]
- Orth, D.; Grif, K.; Khan, A.B.; Naim, A.; Dierich, M.P.; Wurzner, R. The Shiga toxin genotype rather than the amount of Shiga toxin or the cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn. Microbiol. Infect. Dis. 2007, 59, 235–242. [Google Scholar] [CrossRef]
- Ostroff, S.M.; Tarr, P.I.; Neill, M.A.; Lewis, J.H.; Hargrett-Bean, N.; Kobayashi, J.M. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J. Infect. Dis. 1989, 160, 994–998. [Google Scholar] [CrossRef]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [CrossRef]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O’Brien, A.D.; James, M.N. Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [Green Version]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef]
- Sandvig, K.; Bergan, J.; Dyve, A.B.; Skotland, T.; Torgersen, M.L. Endocytosis and retrograde transport of Shiga toxin. Toxicon Off. J. Int. Soc. Toxinol. 2010, 56, 1181–1185. [Google Scholar] [CrossRef]
- Sandvig, K.; Torgersen, M.L.; Engedal, N.; Skotland, T.; Iversen, T.G. Protein toxins from plants and bacteria: Probes for intracellular transport and tools in medicine. FEBS Lett. 2010, 584, 2626–2634. [Google Scholar] [CrossRef]
- Sandvig, K.; Pust, S.; Skotland, T.; van Deurs, B. Clathrin-independent endocytosis: Mechanisms and function. Curr. Opin. Cell Biol. 2011, 23, 413–420. [Google Scholar] [CrossRef]
- Strockbine, N.A.; Marques, L.R.; Holmes, R.K.; O’Brien, A.D. Characterization of monoclonal antibodies against Shiga-like toxin from Escherichia coli. Infect. Immun. 1985, 50, 695–700. [Google Scholar] [CrossRef]
- Perera, L.P.; Marques, L.R.; O’Brien, A.D. Isolation and characterization of monoclonal antibodies to Shiga-like toxin II of enterohemorrhagic Escherichia coli and use of the monoclonal antibodies in a colony enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1988, 26, 2127–2131. [Google Scholar] [CrossRef]
- Mukherjee, J.; Chios, K.; Fishwild, D.; Hudson, D.; O’Donnell, S.; Rich, S.M.; Donohue-Rolfe, A.; Tzipori, S. Production and characterization of protective human antibodies against Shiga toxin 1. Infect. Immun. 2002, 70, 5896–5899. [Google Scholar] [CrossRef]
- Mukherjee, J.; Chios, K.; Fishwild, D.; Hudson, D.; O’Donnell, S.; Rich, S.M.; Donohue-Rolfe, A.; Tzipori, S. Human Stx2-specific monoclonal antibodies prevent systemic complications of Escherichia coli O157:H7 infection. Infect. Immun. 2002, 70, 612–619. [Google Scholar] [CrossRef]
- Sheoran, A.S.; Chapman-Bonofiglio, S.; Harvey, B.R.; Mukherjee, J.; Georgiou, G.; Donohue-Rolfe, A.; Tzipori, S. Human antibody against shiga toxin 2 administered to piglets after the onset of diarrhea due to Escherichia coli O157:H7 prevents fatal systemic complications. Infect. Immun. 2005, 73, 4607–4613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyoshi, D.E.; Rich, C.M.; O’Sullivan-Murphy, S.; Richard, L.; Dilo, J.; Donohue-Rolfe, A.; Sheoran, A.S.; Chapman-Bonofiglio, S.; Tzipori, S. Characterization of a human monoclonal antibody against Shiga toxin 2 expressed in Chinese hamster ovary cells. Infect. Immun. 2005, 73, 4054–4061. [Google Scholar] [CrossRef] [PubMed]
- Pluckthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Bakker, T.R.; Phillips, D.J.; Cornelius, A.; Zitt, C.; Gottler, T.; Sigrist, G.; Fiedler, U.; Ekawardhani, S.; Dolado, I.; et al. Design and characterization of MP0250, a tri-specific anti-HGF/anti-VEGF DARPin(R) drug candidate. MAbs 2017, 9, 1262–1269. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Dawson, K.M.; Binz, H.K. Beyond Antibodies: The DARPin((R)) Drug Platform. BioDrugs 2020, 34, 423–433. [Google Scholar] [CrossRef]
- Steiner, D.; Werz, F.W.; Sonderegger, I.; Goulotti-Georgieva, M.; Villemagne, D.; Phillips, D.J.; Forrer, P.; Stumpp, M.T.; Zitt, C.; Binz, H.K. Half-life extension using serum albumin-binding DARPin domains. Protein Eng. Des. Sel. 2017, 30, 1. [Google Scholar] [CrossRef]
- Simeon, R.; Jiang, M.; Chamoun-Emanuelli, A.M.; Yu, H.; Zhang, Y.; Meng, R.; Peng, Z.; Jakana, J.; Zhang, J.; Feng, H.; et al. Selection and characterization of ultrahigh potency designed ankyrin repeat protein inhibitors of C. difficile toxin B. PLoS Biol. 2019, 17, e3000311. [Google Scholar] [CrossRef]
- Peng, Z.; Simeon, R.; Mitchell, S.B.; Zhang, J.; Feng, H.; Chen, Z. Designed Ankyrin Repeat Protein (DARPin) Neutralizers of TcdB from Clostridium difficile Ribotype 027. mSphere 2019, 4, e00596-19. [Google Scholar] [CrossRef]
- Jermutus, L.; Honegger, A.; Schwesinger, F.; Hanes, J.; Pluckthun, A. Tailoring in vitro evolution for protein affinity or stability. Proc. Natl. Acad. Sci. USA 2001, 98, 75–80. [Google Scholar] [CrossRef]
- Zahnd, C.; Spinelli, S.; Luginbuhl, B.; Amstutz, P.; Cambillau, C.; Pluckthun, A. Directed in vitro evolution and crystallographic analysis of a peptide-binding single chain antibody fragment (scFv) with low picomolar affinity. J. Biol. Chem. 2004, 279, 18870–18877. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Tremblay, J.M.; Mukherjee, J.; Leysath, C.E.; Debatis, M.; Ofori, K.; Baldwin, K.; Boucher, C.; Peters, R.; Beamer, G.; Sheoran, A.; et al. A single VHH-based toxin-neutralizing agent and an effector antibody protect mice against challenge with Shiga toxins 1 and 2. Infect. Immun 2013, 81, 4592–4603. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wiesmann, C.; Fuh, G.; Li, B.; Christinger, H.W.; McKay, P.; de Vos, A.M.; Lowman, H.B. Selection and analysis of an optimized anti-VEGF antibody: Crystal structure of an affinity-matured Fab in complex with antigen. J. Mol. Biol. 1999, 293, 865–881. [Google Scholar] [CrossRef]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [CrossRef]
- Tam, P.J.; Lingwood, C.A. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 2007, 153, 2700–2710. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.K.; O’Brien, A.D.; Ackerman, E.J. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28 S RNA when microinjected into Xenopus oocytes. J. Biol. Chem. 1989, 264, 596–601. [Google Scholar] [CrossRef]
- Zahnd, C.; Kawe, M.; Stumpp, M.T.; de Pasquale, C.; Tamaskovic, R.; Nagy-Davidescu, G.; Dreier, B.; Schibli, R.; Binz, H.K.; Waibel, R.; et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: Effects of affinity and molecular size. Cancer Res. 2010, 70, 1595–1605. [Google Scholar] [CrossRef]
- Sodha, S.V.; Heiman, K.; Gould, L.H.; Bishop, R.; Iwamoto, M.; Swerdlow, D.L.; Griffin, P.M. National patterns of Escherichia coli O157 infections, USA, 1996–2011. Epidemiol. Infect. 2015, 143, 267–273. [Google Scholar] [CrossRef]
- Lupindu, A.M.; Olsen, J.E.; Ngowi, H.A.; Msoffe, P.L.M.; Mtambo, M.M.; Scheutz, F.; Dalsgaard, A. Occurrence and Characterization of Shiga Toxin-Producing Escherichia coli O157:H7 and Other Non-Sorbitol-Fermenting E. coli in Cattle and Humans in Urban Areas of Morogoro, Tanzania. Vector-Borne Zoonot 2014, 14, 503–510. [Google Scholar] [CrossRef]
- Lupindu, A.M. Epidemiology of Shiga toxin-producing Escherichia coli O157:H7 in Africa in review. South Afr. J. Infect. D 2018, 33, 24–30. [Google Scholar] [CrossRef]
- Melton-Celsa, A.R.; O’Brien, A.D. New Therapeutic Developments against Shiga Toxin-Producing Escherichia coli. Microbiol. Spectr. 2014, 2, 2. [Google Scholar] [CrossRef]
- Karmali, M.A.; Steele, B.T.; Petric, M.; Lim, C. Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet 1983, 1, 619–620. [Google Scholar] [CrossRef]
- O’Brien, A.O.; Lively, T.A.; Chen, M.E.; Rothman, S.W.; Formal, S.B. Escherichia coli O157:H7 strains associated with haemorrhagic colitis in the United States produce a Shigella dysenteriae 1 (SHIGA) like cytotoxin. Lancet 1983, 1, 702. [Google Scholar] [CrossRef]
- Corrigan, J.J., Jr.; Boineau, F.G. Hemolytic-uremic syndrome. Pediatr. Rev. 2001, 22, 365–369. [Google Scholar] [CrossRef]
- Smith, M.J.; Melton-Celsa, A.R.; Sinclair, J.F.; Carvalho, H.M.; Robinson, C.M.; O’Brien, A.D. Monoclonal antibody 11E10, which neutralizes shiga toxin type 2 (Stx2), recognizes three regions on the Stx2 A subunit, blocks the enzymatic action of the toxin in vitro, and alters the overall cellular distribution of the toxin. Infect. Immun. 2009, 77, 2730–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpp, M.T.; Forrer, P.; Binz, H.K.; Pluckthun, A. Designing repeat proteins: Modular leucine-rich repeat protein libraries based on the mammalian ribonuclease inhibitor family. J. Mol. Biol. 2003, 332, 471–487. [Google Scholar] [CrossRef]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Pluckthun, A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Lo, A.W.; Moonens, K.; De Kerpel, M.; Brys, L.; Pardon, E.; Remaut, H.; De Greve, H. The molecular mechanism of Shiga toxin Stx2e neutralization by a single-domain antibody targeting the cell receptor-binding domain. J. Biol. Chem. 2014, 289, 25374–25381. [Google Scholar] [CrossRef] [PubMed]
- Mallick, E.M.; McBee, M.E.; Vanguri, V.K.; Melton-Celsa, A.R.; Schlieper, K.; Karalius, B.J.; O’Brien, A.D.; Butterton, J.R.; Leong, J.M.; Schauer, D.B. A novel murine infection model for Shiga toxin-producing Escherichia coli. J. Clin. Investig. 2012, 122, 4012–4024. [Google Scholar] [CrossRef]
- Russo, L.M.; Melton-Celsa, A.R.; Smith, M.A.; Smith, M.J.; O’Brien, A.D. Oral intoxication of mice with Shiga toxin type 2a (Stx2a) and protection by anti-Stx2a monoclonal antibody 11E10. Infect. Immun. 2014, 82, 1213–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Y.; Jiang, M.; Robinson, S.; Peng, Z.; Chonira, V.; Simeon, R.; Tzipori, S.; Zhang, J.; Chen, Z. A Multi-Specific DARPin Potently Neutralizes Shiga Toxin 2 via Simultaneous Modulation of Both Toxin Subunits. Bioengineering 2022, 9, 511. https://doi.org/10.3390/bioengineering9100511
Zeng Y, Jiang M, Robinson S, Peng Z, Chonira V, Simeon R, Tzipori S, Zhang J, Chen Z. A Multi-Specific DARPin Potently Neutralizes Shiga Toxin 2 via Simultaneous Modulation of Both Toxin Subunits. Bioengineering. 2022; 9(10):511. https://doi.org/10.3390/bioengineering9100511
Chicago/Turabian StyleZeng, Yu, Mengqiu Jiang, Sally Robinson, Zeyu Peng, Vikas Chonira, Rudo Simeon, Saul Tzipori, Junjie Zhang, and Zhilei Chen. 2022. "A Multi-Specific DARPin Potently Neutralizes Shiga Toxin 2 via Simultaneous Modulation of Both Toxin Subunits" Bioengineering 9, no. 10: 511. https://doi.org/10.3390/bioengineering9100511