
Thermo-Viscoelastic Response of Protein-Based Hydrogels


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- Polymer chains in protein and peptide gels are connected by (i) temporary bonds between the secondary structures of their blocks (specific protein–protein and protein–peptide interactions [46]), and (ii) covalent cross-links between functional groups of amino acids [47] (which implies that Equation (1) is to be modified to account for the presence of permanent bonds between chains).
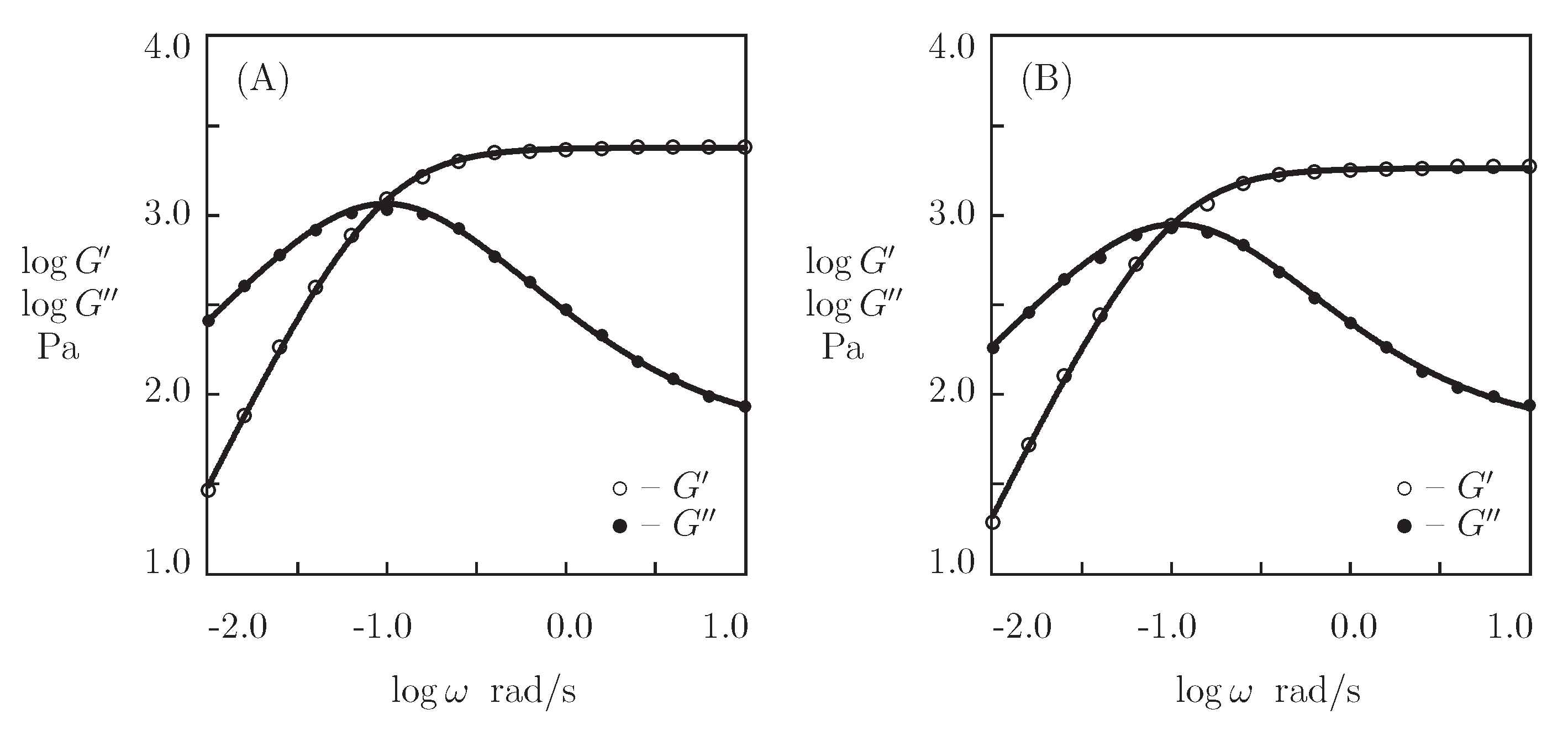
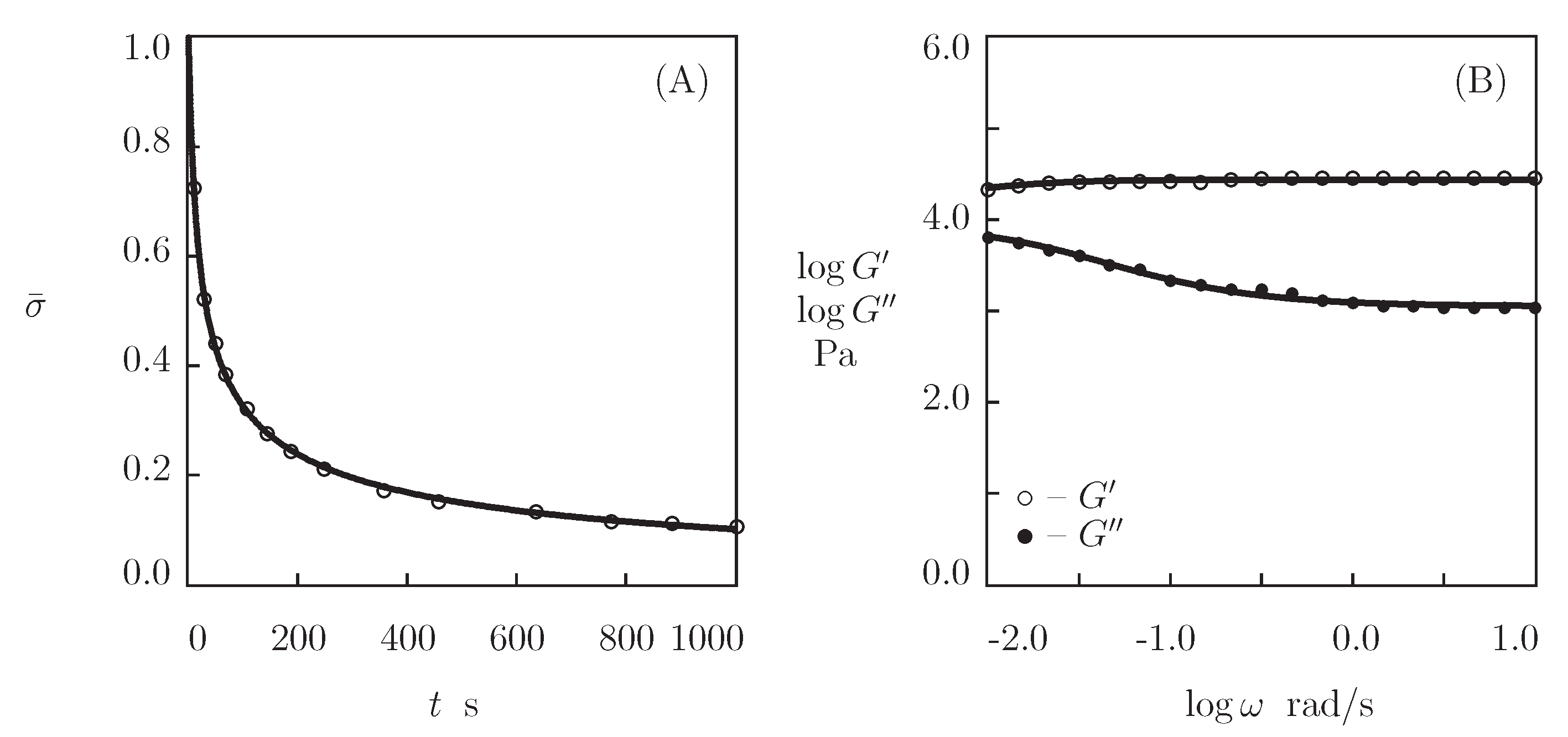
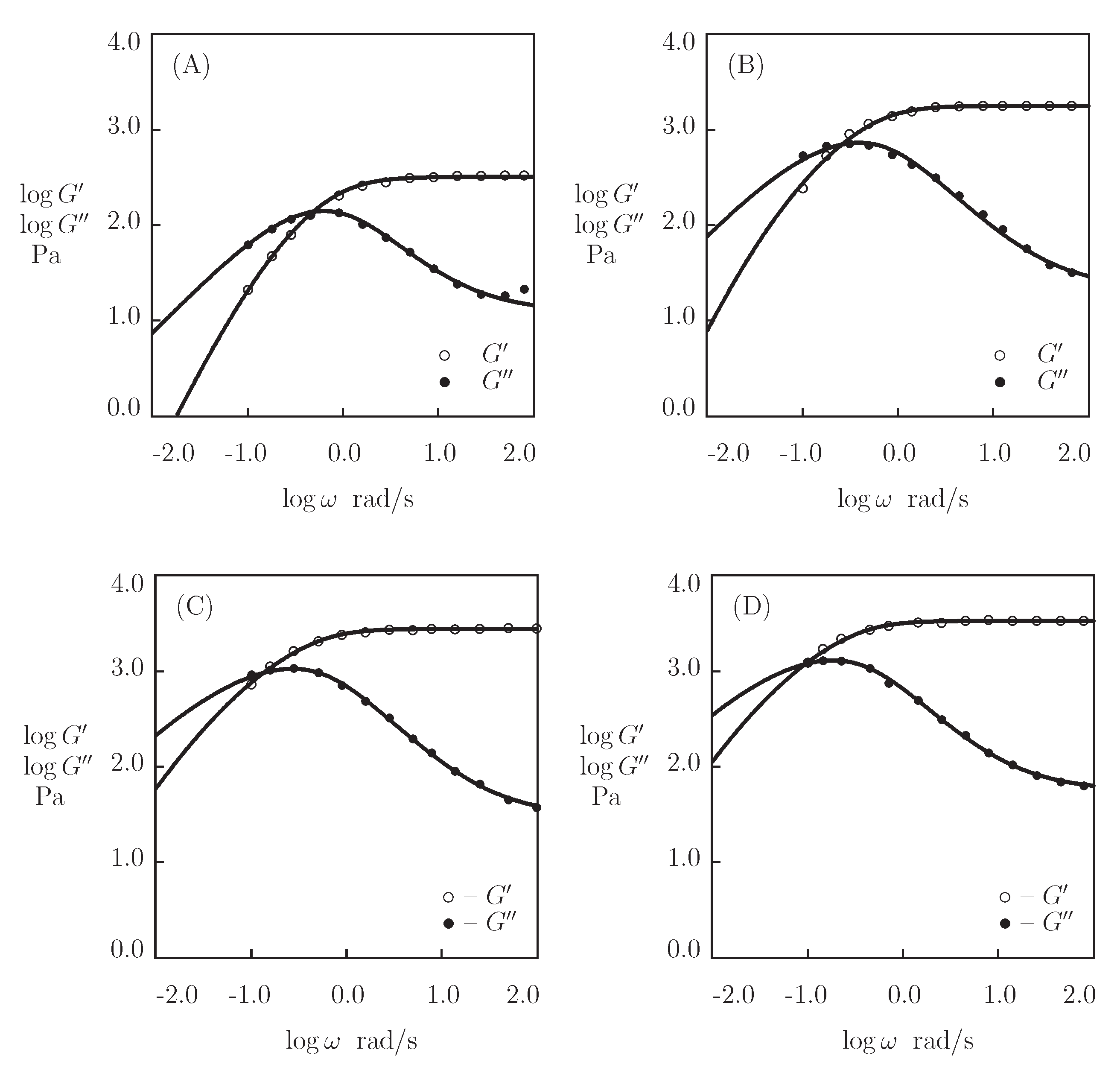
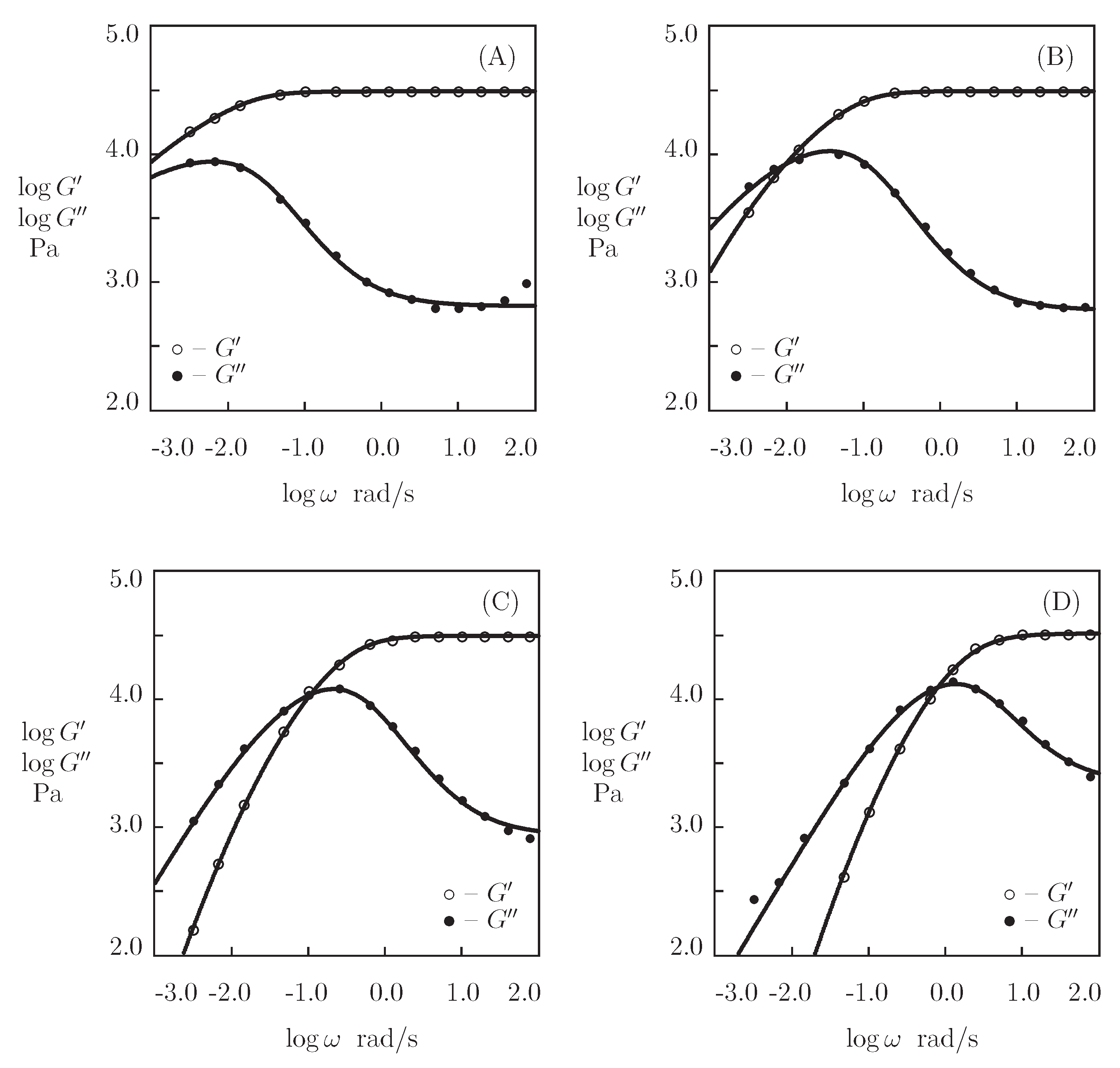
- When the physical cross-links between protein and peptide motifs are sufficiently strong and their rates of dissociation are low under physiological conditions (which implies that the moduli and become practically independent of in the conventional interval of frequencies between 0.1 and 100 rad/s [48]), the kinetics of dissociation of temporary bonds is evaluated by combining the experimental data in shear oscillatory tests, on the one hand, and shear relaxation [49] or creep [50] tests, on the other.
2. Model
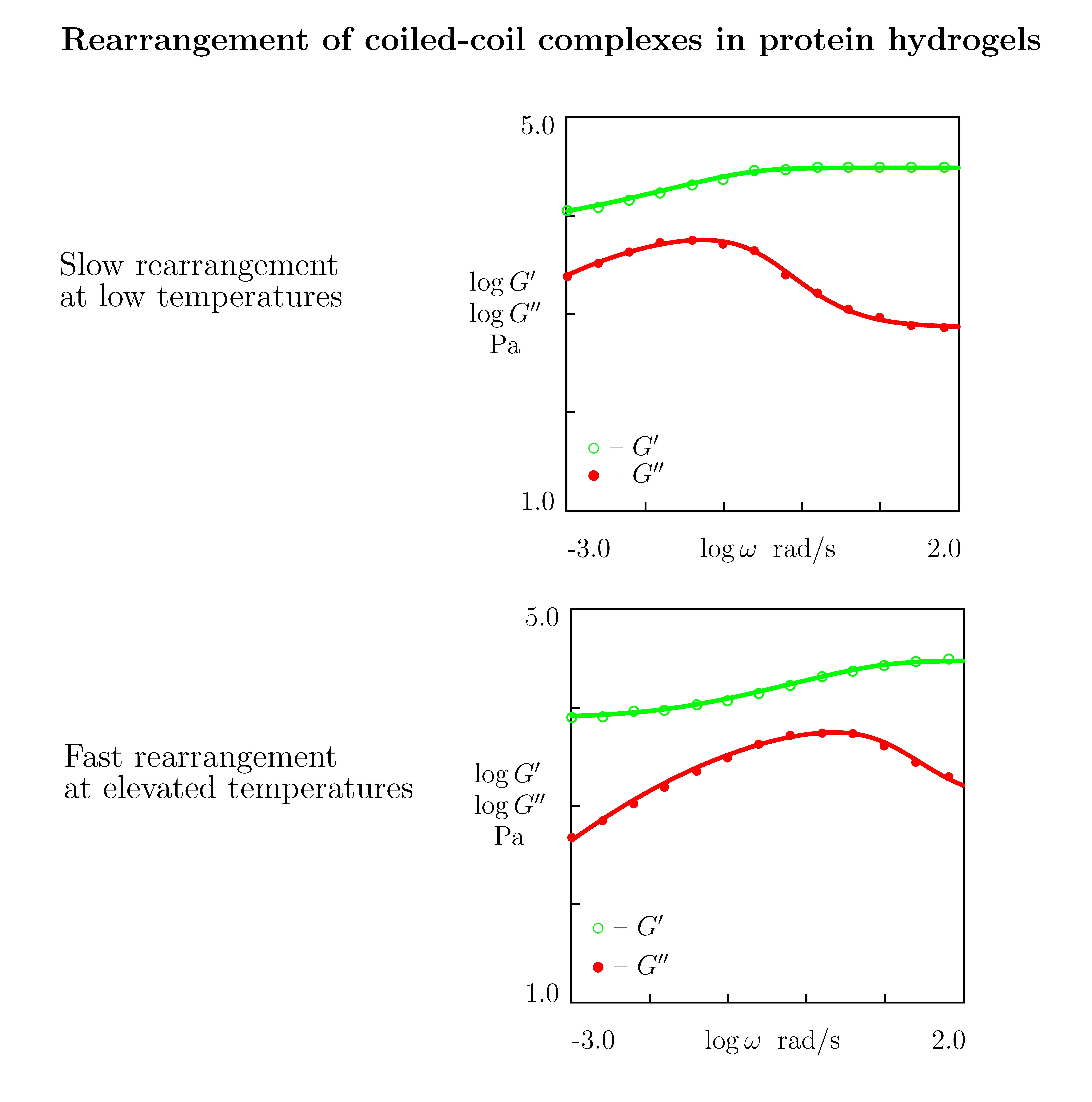
3. Fitting of Experimental Data
4. Biocompatible Supramolecular Gels
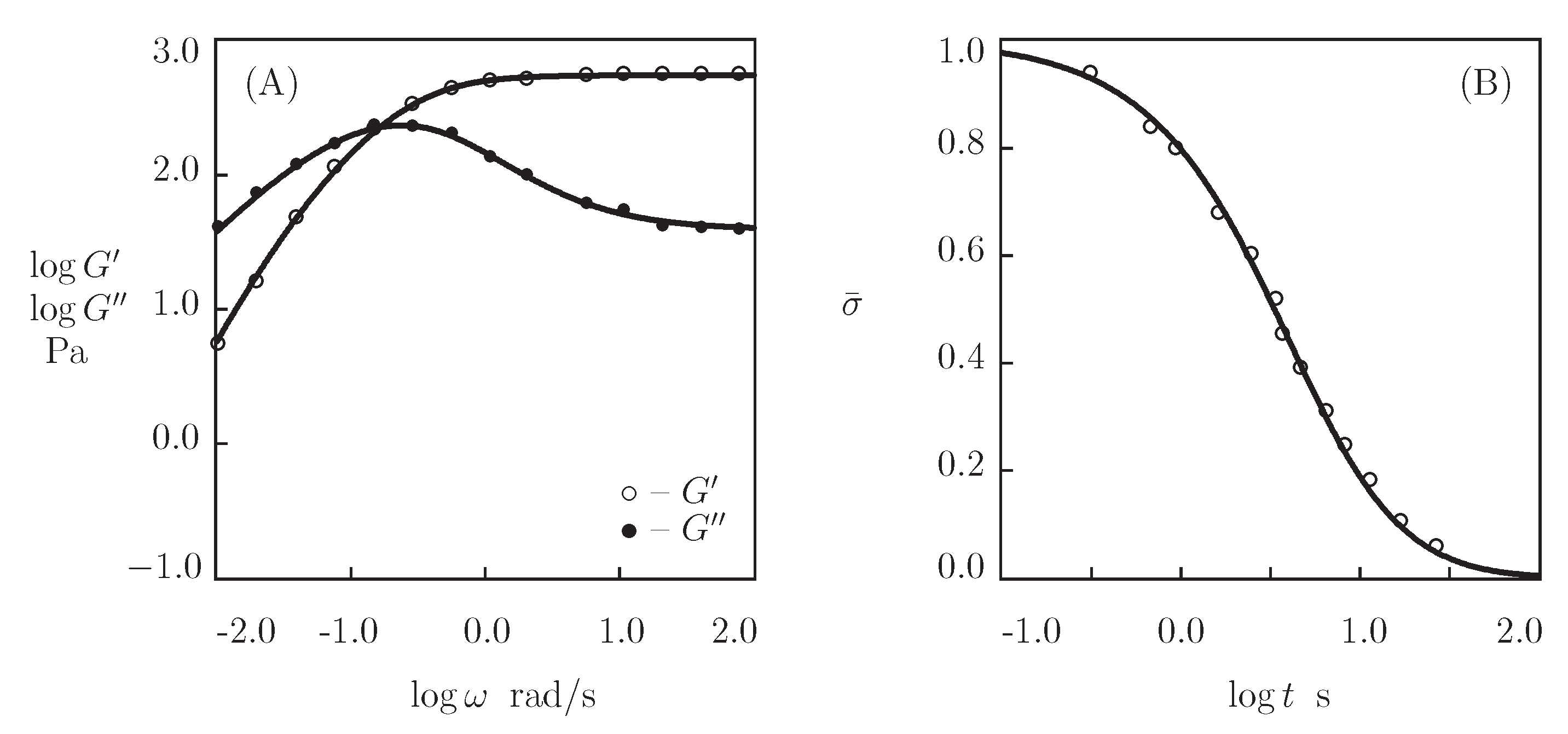
4.1. HA Gel Cross-Linked by Benzoxaborin-Saccharide Complexation
4.2. PEG Gels Cross-Linked by Benzaldehyde-Thiol Complexation
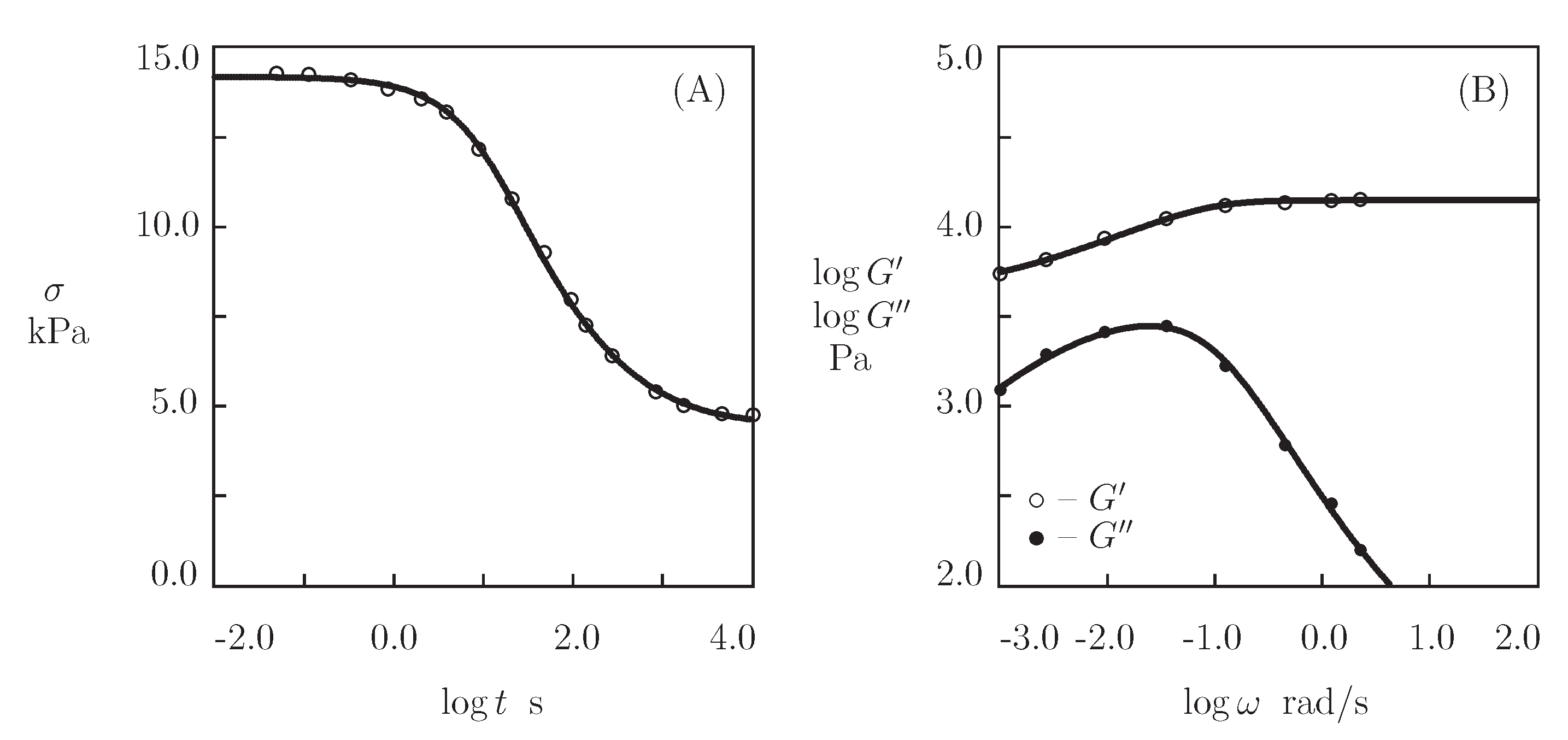
4.3. HA Gels Cross-Linked by Hydrazine-Aldehyde Complexation
4.4. PEG-DDI Gel with Hydrophobic Interactions between Fatty Acids
4.5. Discussion
5. Protein-Based Gels
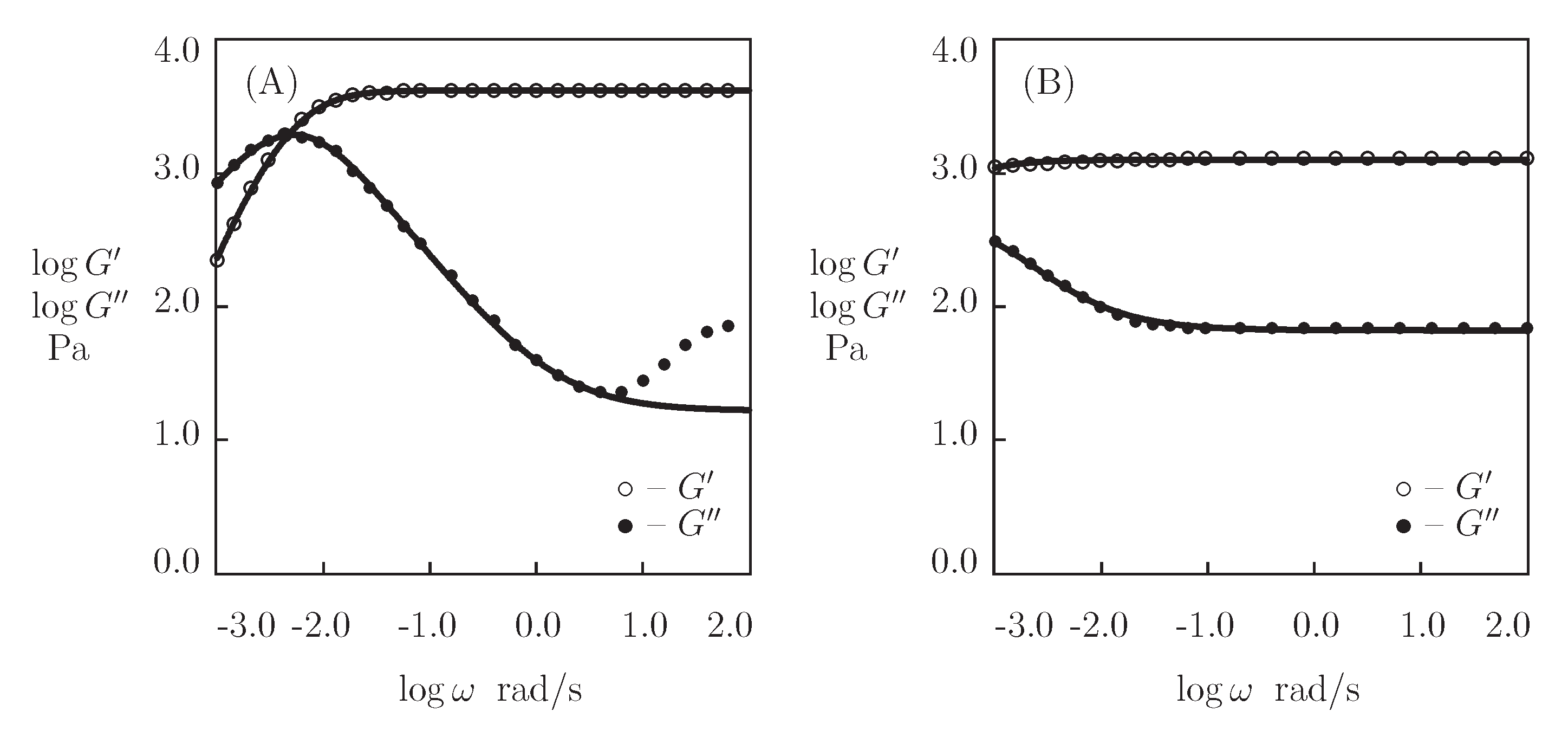
5.1. Peptide-Functionalized PEG Gel
5.2. Protein-Functionalized PEG Gels
5.3. The Effect of Chemical Structure
5.4. The Effect of Concentration
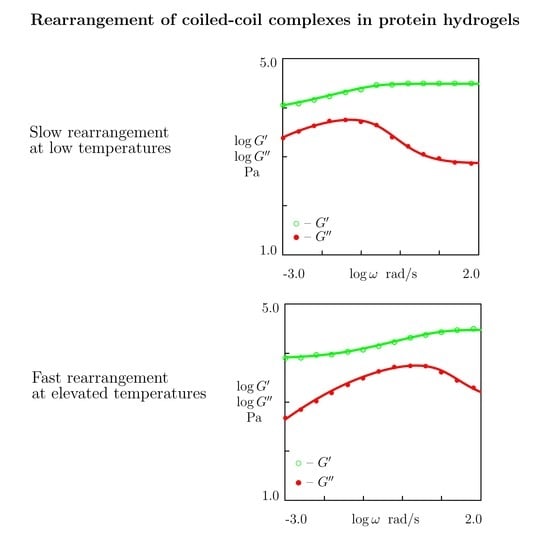
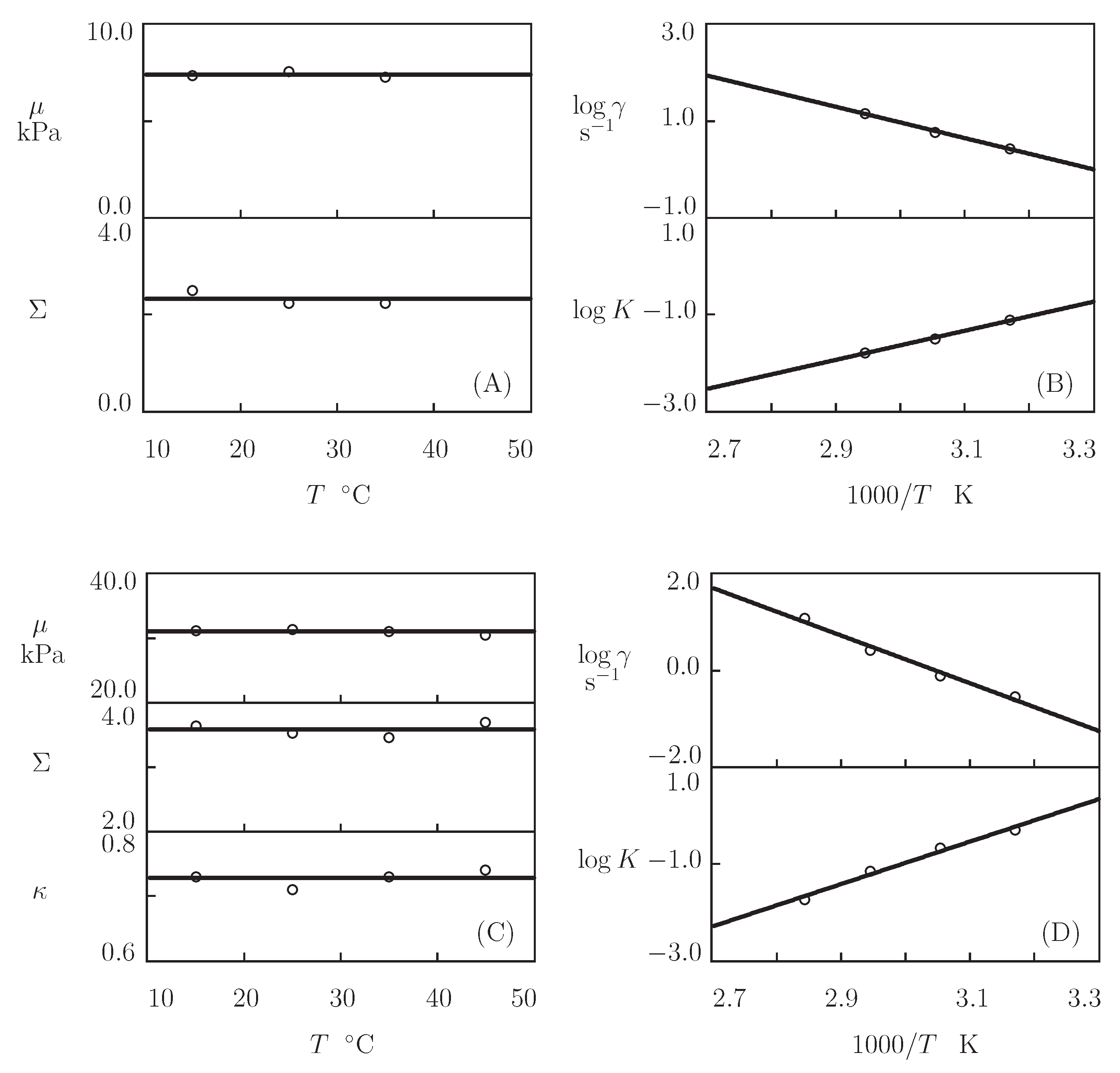
5.5. The Effect of Temperature
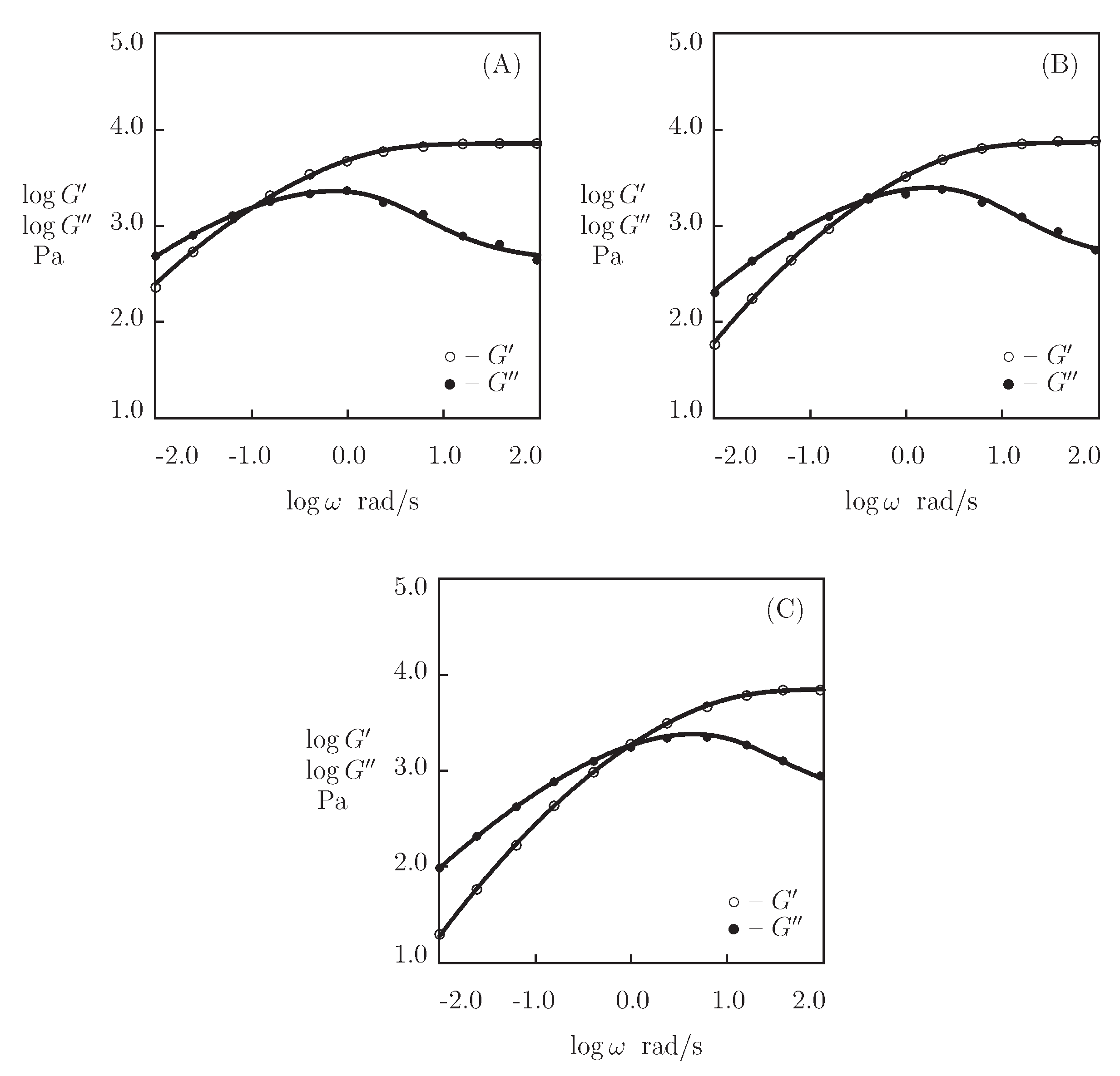
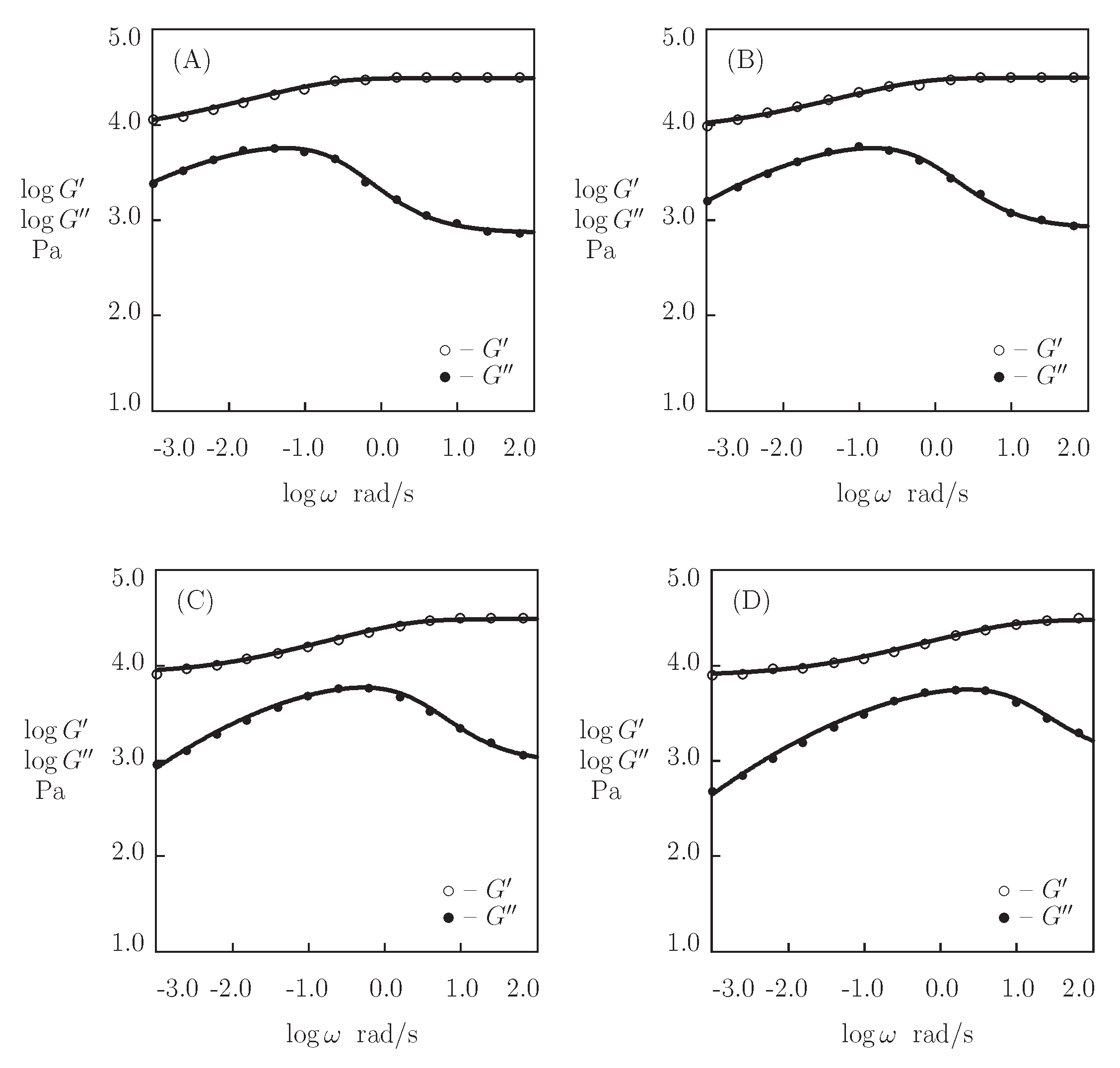
5.6. Discussion
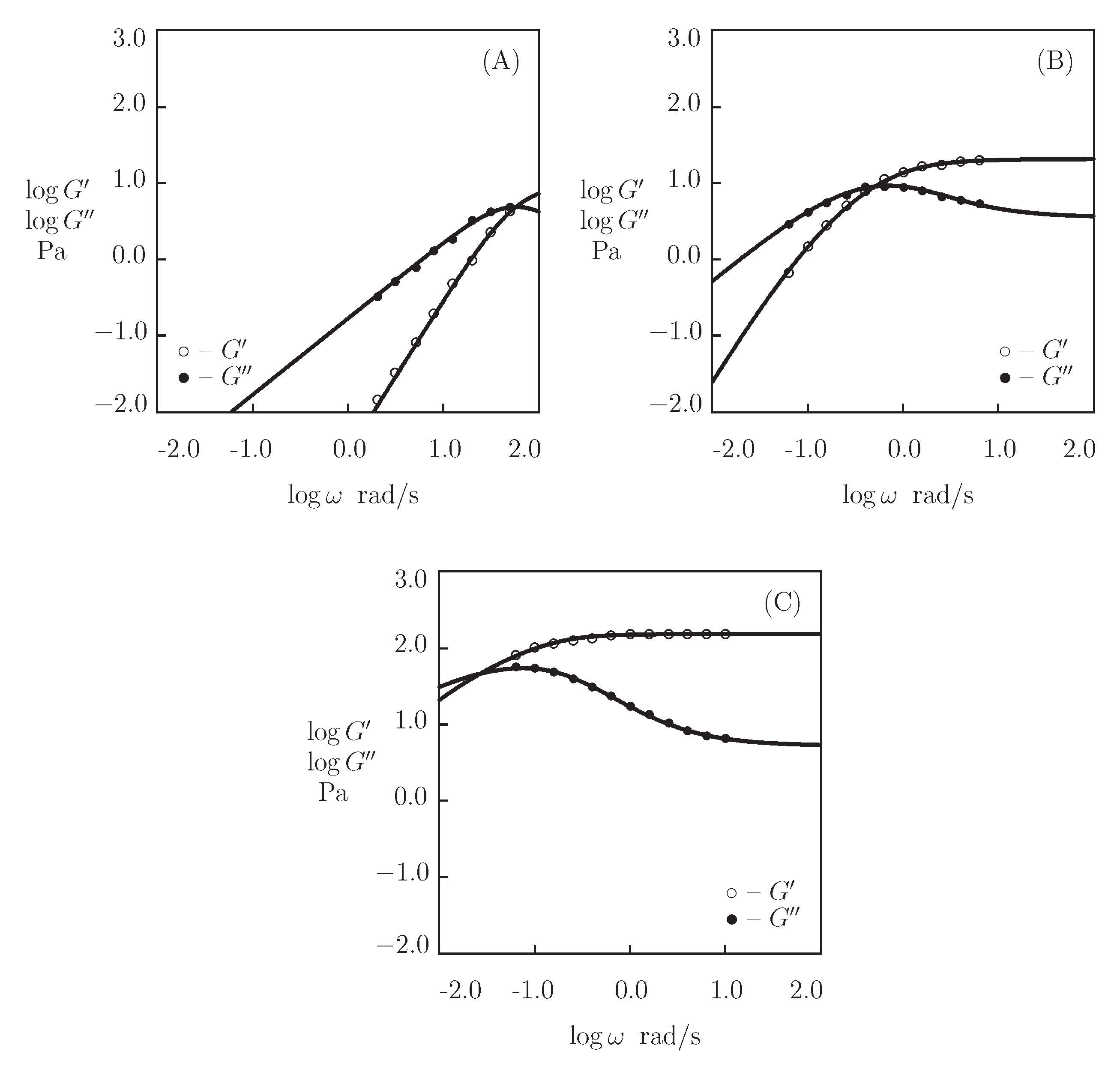
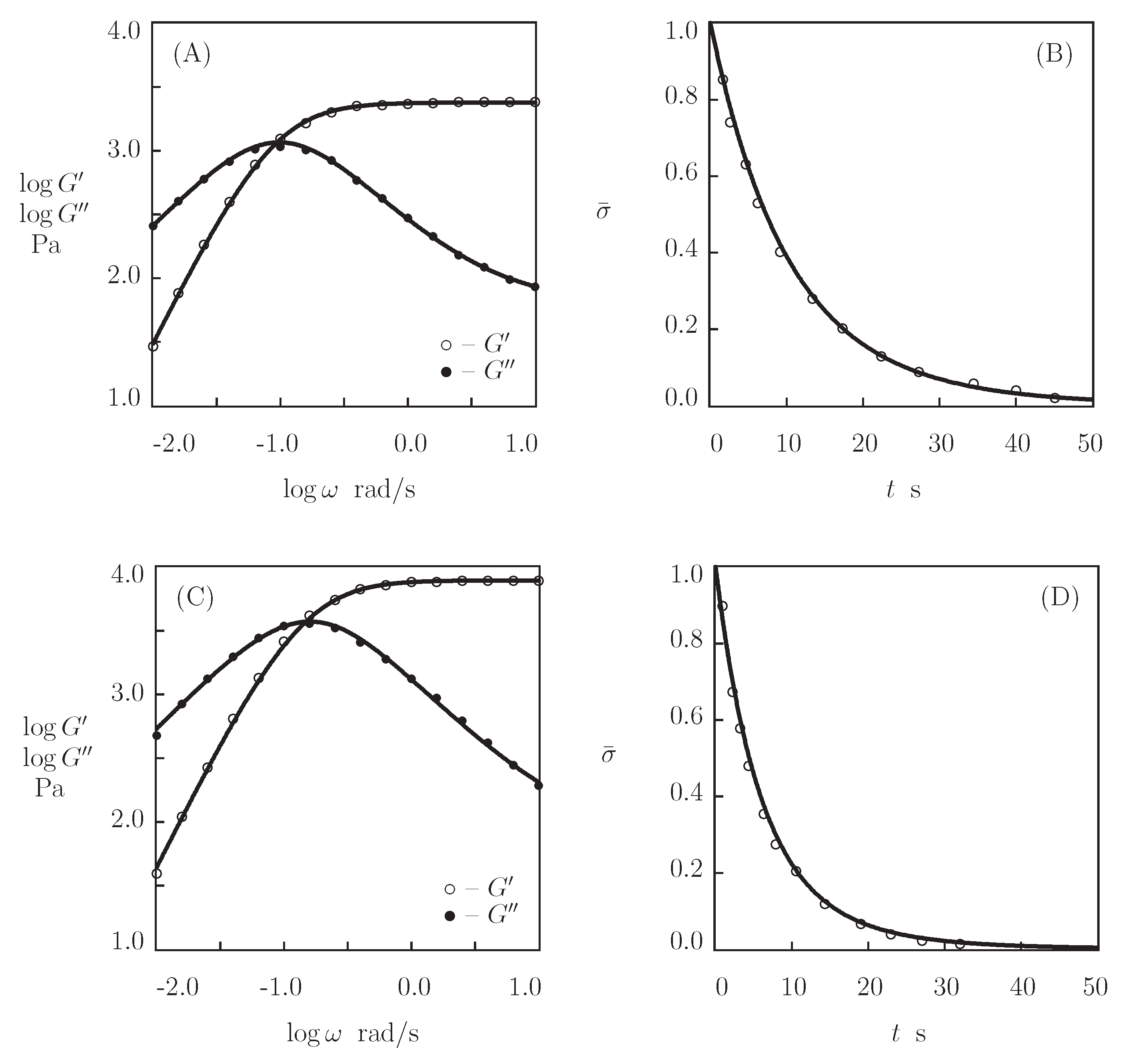
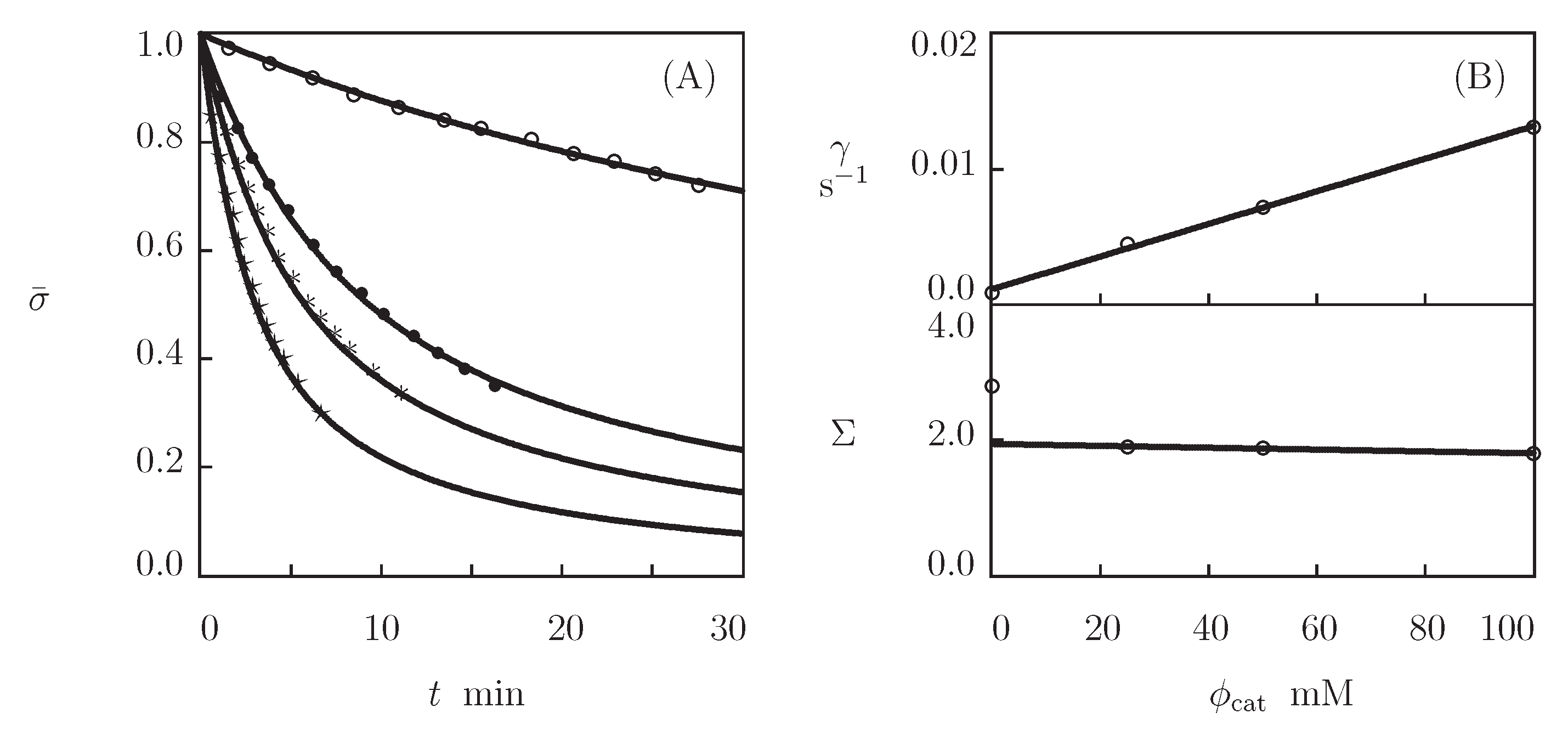
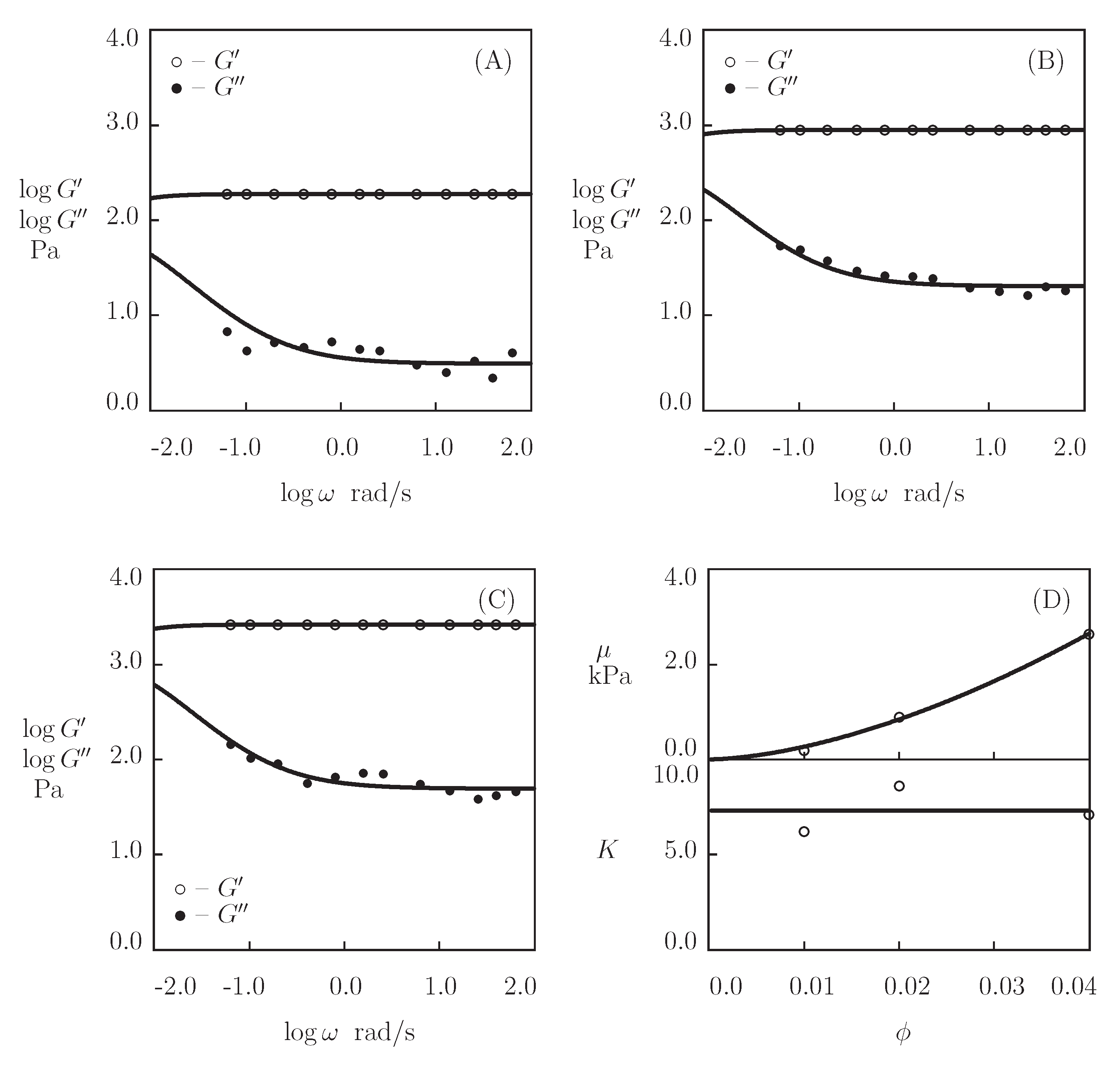
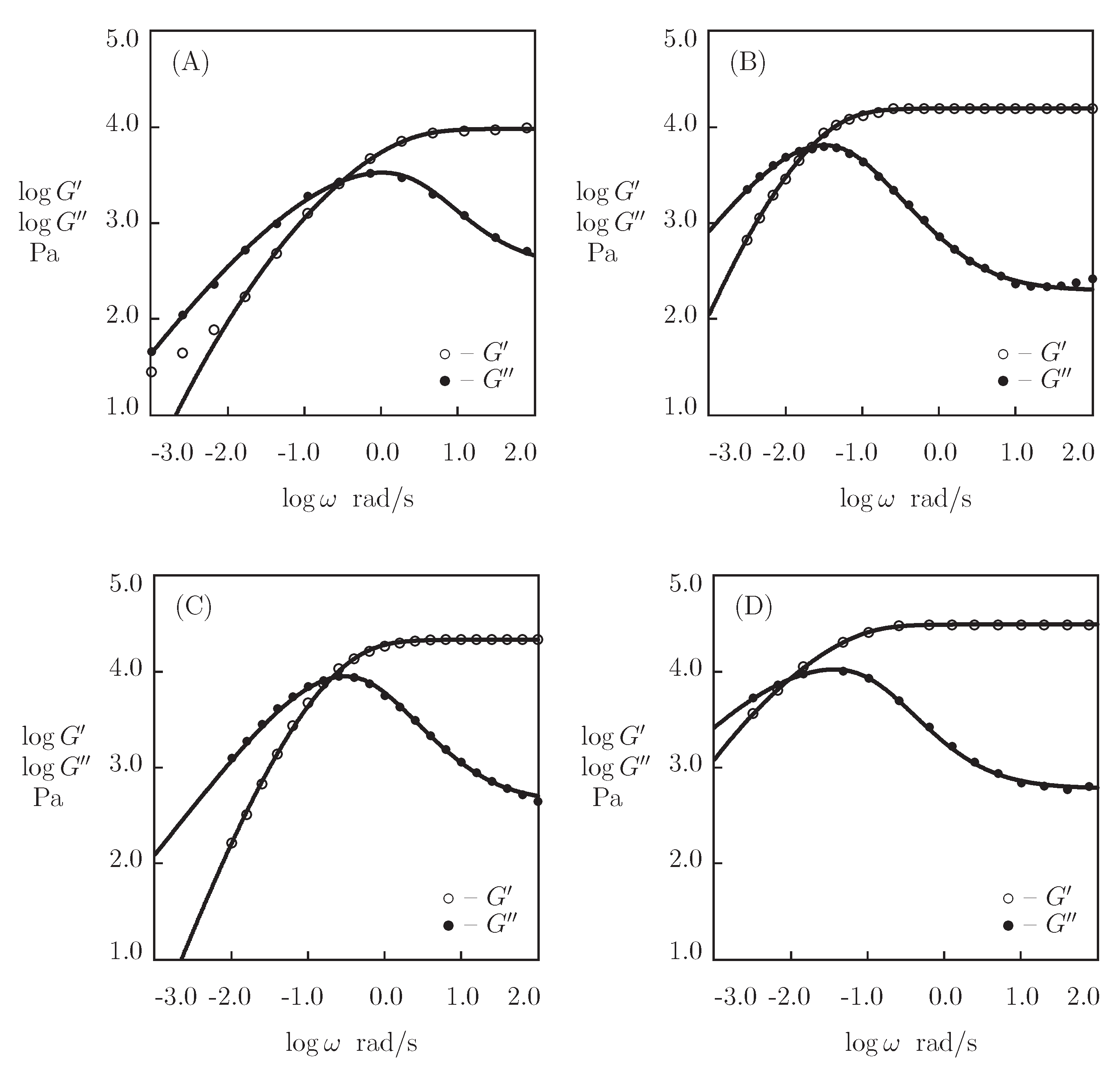
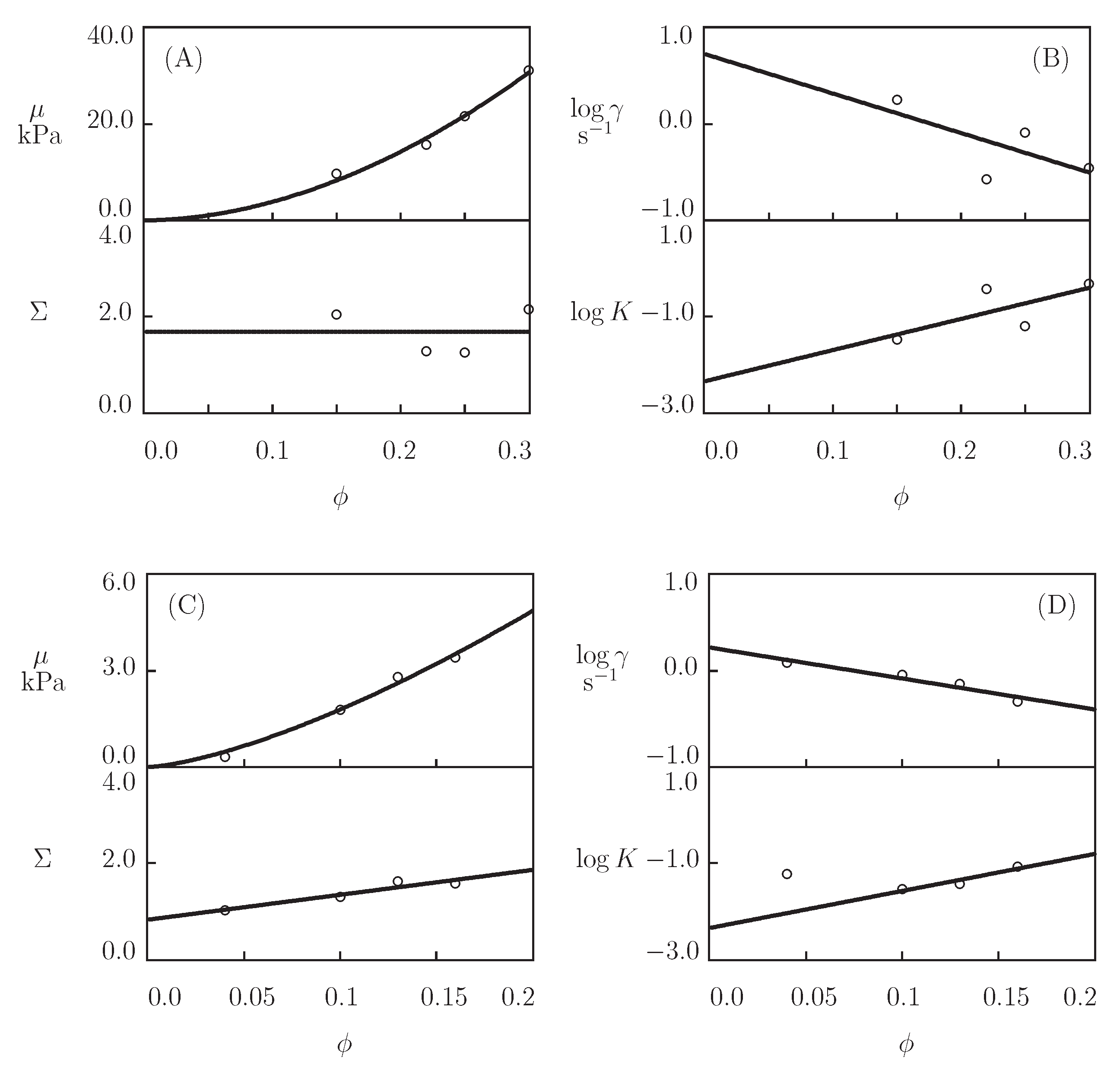
- Figure 13A and Figure 18A,C show that an increase in the concentration of proteins in pre-gel solutions induces a strong increase in the elastic modulus described by Equations (20) and (21) with the exponent m ranging from 1.4 to 1.9. This increase is accompanied by a linear growth of heterogeneity of the network (that is characterized by the coefficient ).
- Figure 13B and Figure 18B,D demonstrate that the growth of induces a pronounced decrease in the rate of dissociation of coiled coil complexes and an increase in the coefficient K (that reflects the influence of disentanglement of chains on the rearrangement of physical bonds). These changes (as described by Equation (21)) may be explained by the strengthening of interactions between coiled coil complexes that are located in close vicinity of each other (these interactions slow down the rearrangement process) and the enhancement of the effect of disentanglement caused by the growth of the effect of disentanglement of concentration of chains.
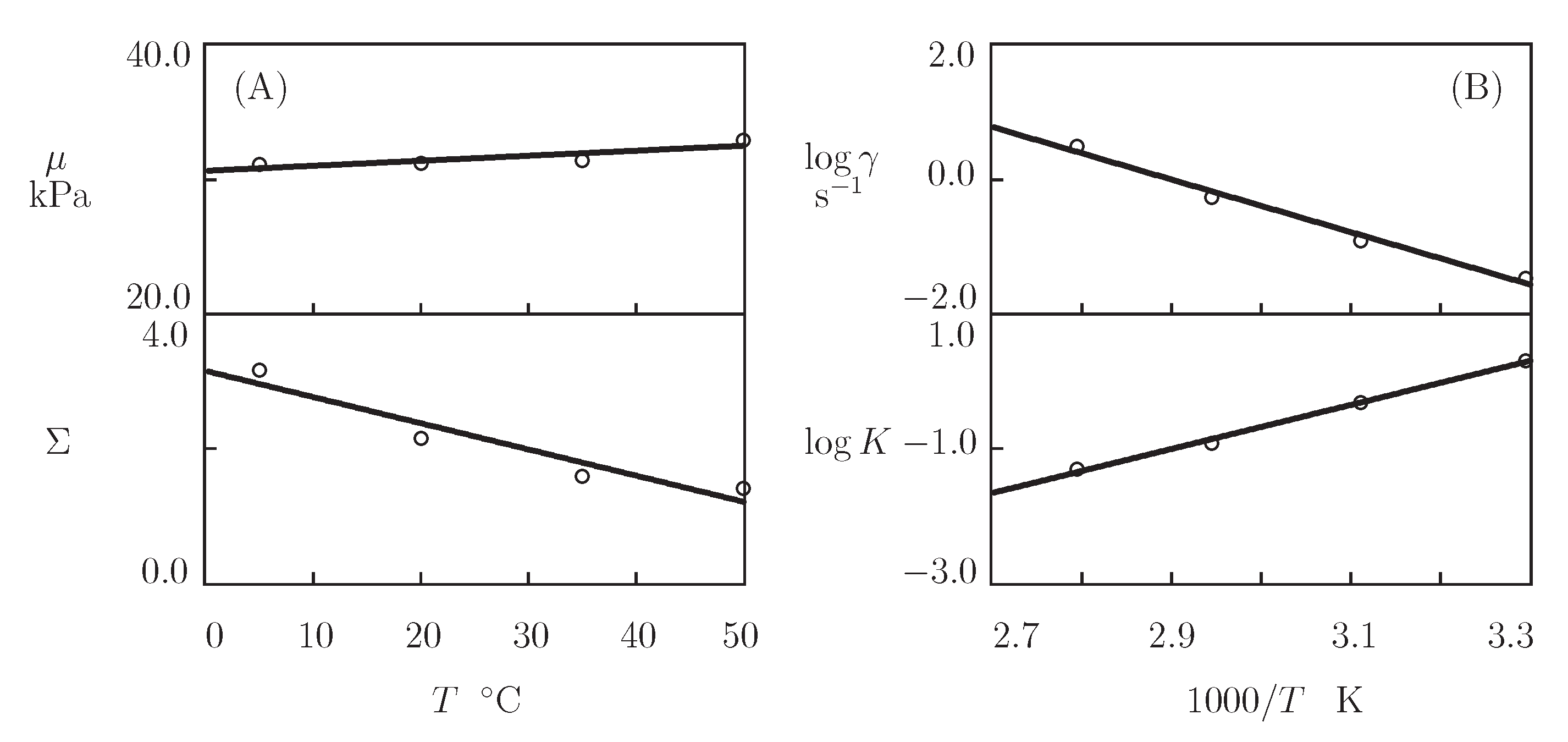
- Figure 15A and Figure 18A,C reveal that an increase in temperature T does not substantially affect the elastic modulus (that is treated as a measure of the concentration of physical bonds between chains), but it can induce a pronounced growth of their strength (that is modeled as a transformation of some temporary bonds into permanent), accompanied by a decrease in the measure of inhomogeneity of the polymer network .
- Figure 15B and Figure 18B,D show that these changes in the network structure occur together with an increase in the rate of dissociation of bonds and a reduction in the coefficient K. Arrhenius Equations (23) with similar values of the activation energies and describe the effect of temperature on and K.
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jonker, A.M.; Lowik, D.W.P.M.; Van Hest, J.C.M. Peptide- and protein-based hydrogels. Chem. Mater. 2012, 24, 759–773. [Google Scholar] [CrossRef]
- Frandsen, J.L.; Ghandehari, H. Recombinant protein-based polymers for advanced drug delivery. Chem. Soc. Rev. 2012, 41, 2696–2706. [Google Scholar] [CrossRef]
- Li, H.; Kong, N.; Laver, B.; Liu, J. Hydrogels constructed from engineered proteins. Small 2016, 12, 973–987. [Google Scholar] [CrossRef]
- Wang, Y.; Katyal, P.; Montclare, J.K. Protein-engineered functional materials. Adv. Healthc. Mater. 2019, 8, 1801374. [Google Scholar] [CrossRef]
- Mondal, S.; Das, S.; Nandi, A.K. A review on recent advances in polymer and peptide hydrogels. Soft Matter 2020, 16, 1404–1454. [Google Scholar] [CrossRef]
- Li, Y.; Xue, B.; Cao, Y. 100th anniversary of macromolecular science viewpoint: Synthetic protein hydrogels. ACS Macro Lett. 2020, 9, 512–524. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Gong, C.; Liu, B.; Li, Y.; Wang, L.; Su, Z.; Wei, G. Recent advances in the fabrication, functionalization, and bioapplications of peptide hydrogels. Soft Matter 2020, 16, 10029–10045. [Google Scholar] [CrossRef] [PubMed]
- Ahn, W.; Lee, J.-H.; Kim, S.R.; Lee, J.; Lee, E.J. Designed protein- and peptide-based hydrogels for biomedical sciences. J. Mater. Chem. B 2021, 9, 1919–1940. [Google Scholar] [CrossRef]
- Apostolovic, B.; Danial, M.; Klok, H.-A. Coiled coils: Attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials. Chem. Soc. Rev. 2010, 39, 3541–3575. [Google Scholar] [CrossRef] [PubMed]
- Lapenta, F.; Aupic, J.; Strmsek, Z.; Jerala, R. Coiled coil protein origami: From modular design principles towards biotechnological applications. Chem. Soc. Rev. 2018, 47, 3530–3542. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Champion, J.A. Self-assembled materials made from functional recombinant proteins. Acc. Chem. Res. 2016, 49, 2188–2198. [Google Scholar] [CrossRef]
- Sing, M.K.; Glassman, M.J.; Vronay-Ruggles, X.T.; Burghardt, W.R.; Olsen, B.D. Structure and rheology of dual-associative protein hydrogels under nonlinear shear flow. Soft Matter 2017, 13, 8511–8524. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, S.; Yan, T.; Fan, X.; Li, F.; Yang, X.; Ren, B.; Xu, J.; Liu, J. Injectable and fast self-healing protein hydrogels. Soft Matter 2019, 15, 7583–7589. [Google Scholar] [CrossRef]
- Zhang, Y.; Desai, M.E.; Wang, T.; Lee, S.-W. Elastin-based thermoresponsive shape-memory hydrogels. Biomacromolecules 2020, 21, 1149–1156. [Google Scholar] [CrossRef]
- Sun, J.; Su, J.; Ma, C.; Gostl, R.; Herrmann, A.; Liu, K.; Zhang, H. Fabrication and mechanical properties of engineered protein-based adhesives and fibers. Adv. Mater. 2020, 32, 1906360. [Google Scholar] [CrossRef]
- Gomes, S.; Leonor, I.B.; Mano, J.F.; Reis, R.L.; Kaplan, D.L. Natural and genetically engineered proteins for tissue engineering. Prog. Polym. Sci. 2012, 37, 1–17. [Google Scholar] [CrossRef]
- Le, D.H.T.; Sugawara-Narutaki, A. Elastin-like polypeptides as building motifs toward designing functional nanobiomaterials. Mol. Syst. Des. Eng. 2019, 4, 545–565. [Google Scholar] [CrossRef]
- Romano, N.H.; Sengupta, D.; Chung, C.; Heilshorn, S.C. Protein-engineered biomaterials: Nanoscale mimics of the extracellular matrix. Biochim. Biophys. Acta 2011, 1810, 339–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annabi, N.; Mithieux, S.M.; Camci-Unal, G.; Dokmeci, M.R.; Weiss, A.S.; Khademhosseini, A. Elastomeric recombinant protein-based biomaterials. Biochem. Eng. J. 2013, 77, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Utterstrom, J.; Naeimipour, S.; Selegard, R.; Aili, D. Coiled coil-based therapeutics and drug delivery systems. Adv. Drug Deliv. Rev. 2021, 170, 26–43. [Google Scholar] [CrossRef]
- Kaygisiz, K.; Synatschke, C.V. Materials promoting viral gene delivery. Biomater. Sci. 2020, 8, 6113–6156. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, X.; Liang, G. Peptide-based supramolecular hydrogels for bioimaging applications. Biomater. Sci. 2021, 9, 315–327. [Google Scholar] [CrossRef]
- Pinelli, F.; Magagnin, L.; Rossi, F. Progress in hydrogels for sensing applications: A review. Mater. Today Chem. 2020, 17, 100317. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, J.R.; Waigh, T.A. Electronics of peptide- and protein-based biomaterials. Adv. Colloid Interface Sci. 2021, 287, 102319. [Google Scholar] [CrossRef] [PubMed]
- Tsoras, A.N.; Champion, J.A. Protein and peptide biomaterials for engineered subunit vaccines and immunotherapeutic applications. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 337–359. [Google Scholar] [CrossRef]
- Stern, D.; Cui, H. Crafting polymeric and peptidic hydrogels for improved wound healing. Adv. Healthc. Mater. 2019, 8, 1900104. [Google Scholar] [CrossRef]
- Sato, K.; Hendricks, M.P.; Palmer, L.C.; Stupp, S.I. Peptide supramolecular materials for therapeutics. Chem. Soc. Rev. 2018, 47, 7539–7551. [Google Scholar] [CrossRef]
- Ding, X.; Zhao, H.; Li, Y.; Lee, A.L.; Li, Z.; Fu, M.; Li, C.; Yang, Y.Y.; Yuan, P. Synthetic peptide hydrogels as 3D scaffolds for tissue engineering. Adv. Drug Deliv. Rev. 2020, 160, 78–104. [Google Scholar] [CrossRef]
- Chung, J.; Kushner, A.M.; Weisman, A.C.; Guan, Z. Direct correlation of single-molecule properties with bulk mechanical performance for the biomimetic design of polymers. Nat. Mater. 2014, 13, 1055–1062. [Google Scholar] [CrossRef]
- Wang, R.; Sing, M.K.; Avery, R.K.; Souza, B.S.; Kim, M.; Olsen, B.D. Classical challenges in the physical chemistry of polymer networks and the design of new materials. Acc. Chem. Res. 2016, 49, 2786–2795. [Google Scholar] [CrossRef]
- Gomes, M.C.; Mano, J.F. Chemical modification strategies to prepare advanced protein-based biomaterials. Biomater. Biosyst. 2021, 1, 100010. [Google Scholar] [CrossRef]
- Yan, C.; Pochan, D.J. Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem. Soc. Rev. 2010, 39, 3528–3540. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.H.; Pathiranage, T.M.S.K.; Marciel, A.B. Rheological properties of engineered protein polymer networks. MRS Bull. 2020, 45, 1048–1054. [Google Scholar] [CrossRef]
- Guvendiren, M.; Lu, H.D.; Burdick, J.A. Shear-thinning hydrogels for biomedical applications. Soft Matter 2012, 8, 260–272. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Gu, L.; Klumpers, D.; Darnell, M.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Lee, H.-P.; Lippens, E.; Duda, G.N.; et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 2016, 15, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Dawn, A.; Kumari, H. Low molecular weight supramolecular gels under shear: Rheology as the tool for elucidating structure-function correlation. Chem. Eur. J. 2018, 24, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.L.; Yu, A.C.; Agmon, G.; Appel, E.A. Supramolecular polymeric biomaterials. Biomater. Sci. 2018, 6, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Gonzalez, M.; Willner, I. Stimuli-responsive biomolecule-based hydrogels and their applications. Angew. Chem. Int. Ed. 2020, 59, 15342–15377. [Google Scholar] [CrossRef] [PubMed]
- Grindy, S.C.; Learsch, R.; Mozhdehi, D.; Cheng, J.; Barrett, D.G.; Guan, Z.; Messersmith, P.B.; Holten-Andersen, N. Control of hierarchical polymer mechanics with bioinspired metal-coordination dynamics. Nat. Mater. 2015, 14, 1210–1216. [Google Scholar] [CrossRef]
- Tan, C.S.Y.; Agmon, G.; Liu, J.; Hoogland, D.; Janecek, E.-R.; Appel, E.A.; Scherman, O.A. Distinguishing relaxation dynamics in transiently crosslinked polymeric networks. Polym. Chem. 2017, 8, 5336–5343. [Google Scholar] [CrossRef]
- Ahmadi, M.; Seiffert, S. Thermodynamic control over energy dissipation modes in dual-network hydrogels based on metal-ligand coordination. Soft Matter 2020, 16, 2332–2341. [Google Scholar] [CrossRef]
- Ahmadi, M.; Seiffert, S. Coordination geometry preference regulates the structure and dynamics of metallo-supramolecular polymer networks. Macromolecules 2021, 54, 1388–1400. [Google Scholar] [CrossRef]
- Drozdov, A.D.; deClaville Christiansen, J. Structure–property relations in linear viscoelasticity of supramolecular hydrogels. RSC Adv. 2021, 11, 16860–16880. [Google Scholar] [CrossRef]
- Singh, N.; Kumar, M.; Miravet, J.F.; Ulijn, R.V.; Escuder, B. Peptide-based molecular hydrogels as supramolecular protein mimics. Chem. Eur. J. 2017, 23, 981–993. [Google Scholar] [CrossRef]
- Wang, H.; Shi, Y.; Wang, L.; Yang, Z. Recombinant proteins as cross-linkers for hydrogelations. Chem. Soc. Rev. 2013, 42, 891–901. [Google Scholar] [CrossRef]
- Dooling, L.J.; Tirrell, D.A. Engineering the dynamic properties of protein networks through sequence variation. ACS Cent. Sci. 2016, 2, 812–819. [Google Scholar] [CrossRef]
- Cai, L.; Liu, S.; Guo, J.; Jia, Y.-G. Polypeptide-based self-healing hydrogels: Design and biomedical applications. Acta Biomater. 2020, 113, 84–100. [Google Scholar] [CrossRef]
- Yang, Z.; Kou, S.; Wei, X.; Zhang, F.; Li, F.; Wang, X.-W.; Lin, Y.; Wan, C.; Zhang, W.-B.; Sun, F. Genetically programming stress-relaxation behavior in entirely protein-based molecular networks. ACS Macro Lett. 2018, 7, 1468–1474. [Google Scholar] [CrossRef]
- Aufderhorst-Roberts, A.; Hughes, M.D.G.; Hare, A.; Head, D.A.; Kapur, N.; Brockwell, D.J.; Dougan, L. Reaction rate governs the viscoelasticity and nanostructure of folded protein hydrogels. Biomacromolecules 2020, 21, 4253–4260. [Google Scholar] [CrossRef]
- Green, M.S.; Tobolsky, A.V. A new approach to the theory of relaxing polymeric media. J. Chem. Phys. 1946, 14, 80–92. [Google Scholar] [CrossRef]
- Tanaka, F.; Edwards, S.F. Viscoelastic properties of physically cross-linked networks. Transient network theory. Macromolecules 1992, 25, 1516–1523. [Google Scholar] [CrossRef]
- Derrida, B. Random-energy model: Limit of a family of disordered models. Phys. Rev. Lett. 1980, 45, 79–92. [Google Scholar] [CrossRef]
- Drozdov, A.D. A model for the nonlinear viscoelastic response in polymers at finite strains. Int. J. Solids Struct. 1998, 35, 2315–2347. [Google Scholar] [CrossRef]
- Drozdov, A.D. A constitutive model in finite thermoviscoelasticity based on the concept of transient networks. Acta Mech. 1999, 133, 13–37. [Google Scholar] [CrossRef]
- Guo, J.; Long, P.; Mayumi, K.; Hui, C.-Y. Mechanics of a dual cross-link gel with dynamic bonds: Steady state kinetics and large deformation effects. Macromolecules 2016, 49, 3497–3507. [Google Scholar] [CrossRef]
- Zheng, S.Y.; Liu, C.; Jiang, L.; Lin, J.; Qian, J.; Mayumi, K.; Wu, Z.L.; Ito, K.; Zheng, Q. Slide-ring cross-links mediated tough metallo-supramolecular hydrogels with superior self-recoverability. Macromolecules 2019, 52, 6748–6755. [Google Scholar] [CrossRef]
- Parada, G.A.; Zhao, X. Ideal reversible polymer networks. Soft Matter 2018, 14, 5186–5196. [Google Scholar] [CrossRef] [Green Version]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Klein, J. Evidence for reptation in an entangled polymer melt. Nature 1978, 271, 143–145. [Google Scholar] [CrossRef]
- Drozdov, A.D.; deClaville Christiansen, J. Nanocomposite gels with permanent and transient junctions under cyclic loading. Macromolecules 2018, 51, 1462–1473. [Google Scholar] [CrossRef]
- Jangizehi, A.; Ghaffarian, S.R.; Schmolke, W.; Seiffert, S. Dominance of chain entanglement over transient sticking on chain dynamics in hydrogen-bonded supramolecular polymer networks in the melt. Macromolecules 2018, 51, 2859–2871. [Google Scholar] [CrossRef]
- Drozdov, A.D.; Kalamkarov, A.L. A constitutive model for nonlinear viscoelastic behavior of polymers. Polym. Eng. Sci. 1996, 36, 1907–1919. [Google Scholar] [CrossRef]
- Sim, H.G.; Ahn, K.H.; Lee, S.J. Large amplitude oscillatory shear behavior of complex fluids investigated by a network model: A guideline for classification. J. Non-Newton. Fluid Mech. 2003, 112, 237–250. [Google Scholar] [CrossRef]
- Tripathi, A.; Tam, K.C.; McKinley, G.H. Rheology and dynamics of associative polymers in shear and extension: Theory and experiments. Macromolecules 2006, 39, 1981–1999. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Sun, W.; Fan, Q.; Xue, B.; Li, Y.; Qin, M.; Li, Y.; Chen, B.; Wang, W.; Cao, Y. Tuning of the dynamics of metal ion crosslinked hydrogels by network structures. Soft Matter 2019, 15, 4423–4427. [Google Scholar] [CrossRef]
- Figueiredo, T.; Jing, J.; Jeacomine, I.; Olsson, J.; Gerfaud, T.; Boiteau, J.-G.; Rome, C.; Harris, C.; Auzely-Velty, R. Injectable self-healing hydrogels based on boronate ester formation between hyaluronic acid partners modified with benzoxaborin derivatives and saccharides. Biomacromolecules 2020, 21, 230–239. [Google Scholar] [CrossRef]
- FitzSimons, T.M.; Oentoro, F.; Shanbhag, T.V.; Anslyn, E.V.; Rosales, A.M. Preferential control of forward reaction kinetics in hydrogels crosslinked with reversible conjugate additions. Macromolecules 2020, 53, 3738–3746. [Google Scholar] [CrossRef]
- Lou, J.; Liu, F.; Lindsay, C.D.; Chaudhuri, O.; Heilshorn, S.C.; Xia, Y. Dynamic hyaluronan hydrogels with temporally modulated high injectability and stability using a biocompatible catalyst. Adv. Mater. 2018, 30, 1705215. [Google Scholar] [CrossRef]
- Mihajlovic, M.; Staropoli, M.; Appavou, M.-S.; Wyss, H.M.; Pyckhout-Hintzen, W.; Sijbesma, R.P. Tough supramolecular hydrogel based on strong hydrophobic interactions in a multiblock segmented copolymer. Macromolecules 2017, 50, 3333–3346. [Google Scholar] [CrossRef]
- Tunn, I.; Harrington, M.J.; Blank, K.G. Bioinspired histidine–Zn2+ coordination for tuning the mechanical properties of self-healing coiled coil cross-linked hydrogels. Biomimetics 2019, 4, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, B.D.; Kornfield, J.A.; Tirrell, D.A. Yielding behavior in injectable hydrogels from telechelic proteins. Macromolecules 2010, 43, 9094–9099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassman, M.J.; Chan, J.; Olsen, B.D. Reinforcement of shear thinning protein hydrogels by responsive block copolymer self-assembly. Adv. Funct. Mater. 2013, 23, 1182–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Glassman, M.J.; Li, S.; Socrate, S.; Olsen, B.D. Oxidatively responsive chain extension to entangle engineered protein hydrogels. Macromolecules 2014, 47, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Wang, M.; Olsen, B.D. Anomalous self-diffusion and sticky Rouse dynamics in associative protein hydrogels. J. Am. Chem. Soc. 2015, 137, 3946–3957. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Duan, T.; Cao, Y.; Li, H. An injectable self-healing protein hydrogel with multiple dissipation modes and tunable dynamic response. Biomacromolecules 2019, 20, 4199–4207. [Google Scholar] [CrossRef]
- Rubinstein, M.; Semenov, A.N. Dynamics of entangled solutions of associating polymers. Macromolecules 2001, 34, 1058–1068. [Google Scholar] [CrossRef]
- Yang, M.; Shi, J.; Schlenoff, J.B. Control of dynamics in polyelectrolyte complexes by temperature and salt. Macromolecules 2019, 52, 1930–1941. [Google Scholar] [CrossRef]
- Xian, S.; Webber, M.J. Temperature-responsive supramolecular hydrogels. J. Mater. Chem. B 2020, 8, 9197–9211. [Google Scholar] [CrossRef] [PubMed]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drozdov, A.D.; deClaville Christiansen, J. Thermo-Viscoelastic Response of Protein-Based Hydrogels. Bioengineering 2021, 8, 73. https://doi.org/10.3390/bioengineering8060073
Drozdov AD, deClaville Christiansen J. Thermo-Viscoelastic Response of Protein-Based Hydrogels. Bioengineering. 2021; 8(6):73. https://doi.org/10.3390/bioengineering8060073
Chicago/Turabian StyleDrozdov, Aleksey D., and Jesper deClaville Christiansen. 2021. "Thermo-Viscoelastic Response of Protein-Based Hydrogels" Bioengineering 8, no. 6: 73. https://doi.org/10.3390/bioengineering8060073