
In Silico Analysis of Glucose Oxidase from Aspergillus niger: Potential Cysteine Mutation Sites for Enhancing Protein Stability



Abstract
:1. Introduction
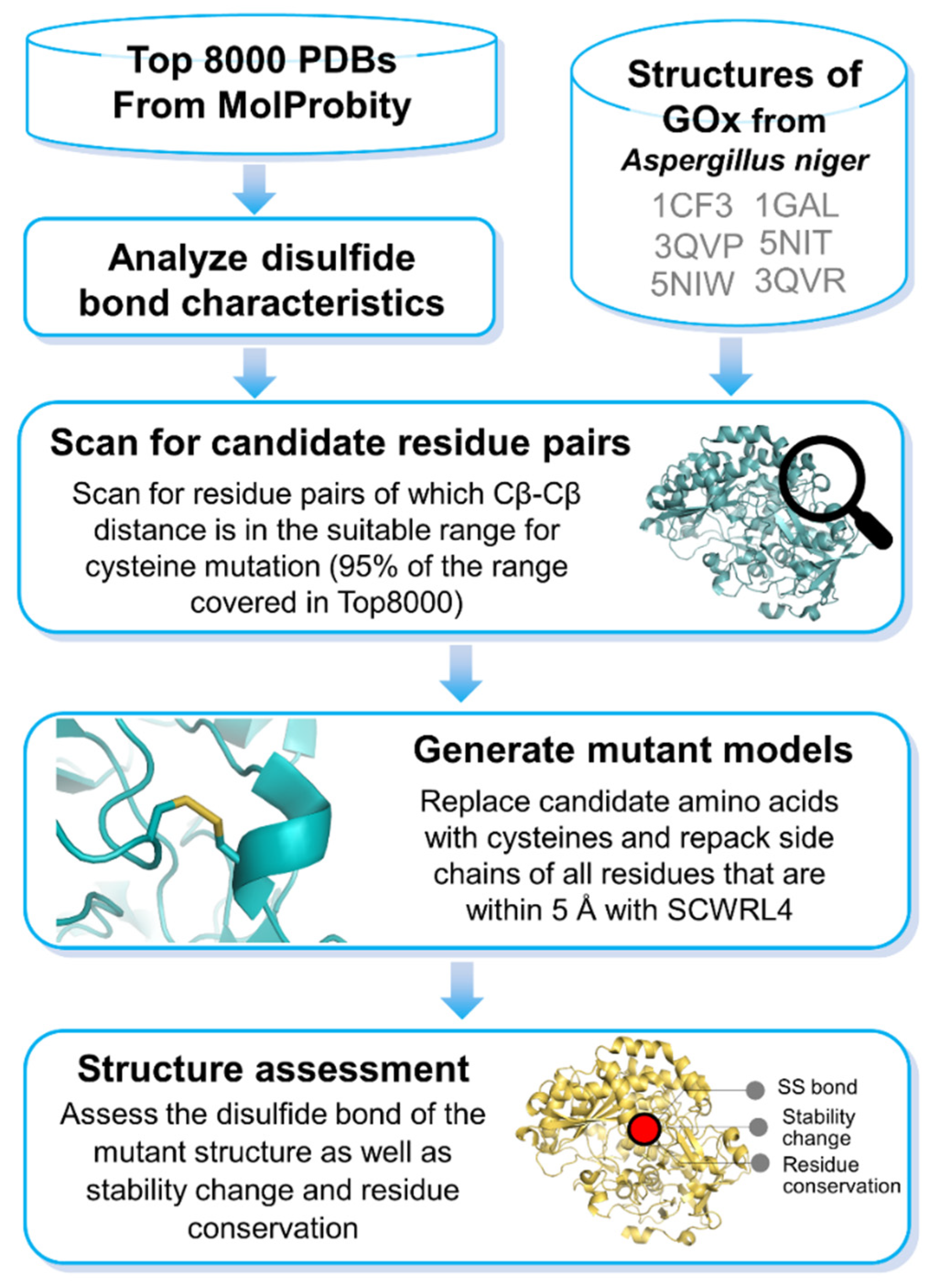
2. Materials and Methods
2.1. Analysis of Disulfide Bonds
2.2. Screening for Candidate Residues in GOx
2.3. Cysteine Repacking and Assessment of Disulfide Bond
2.4. Prediction of Functionally Important Residue and Mutation Effects on GOx
2.5. Prediction of Stability Changes on GOx upon Mutations
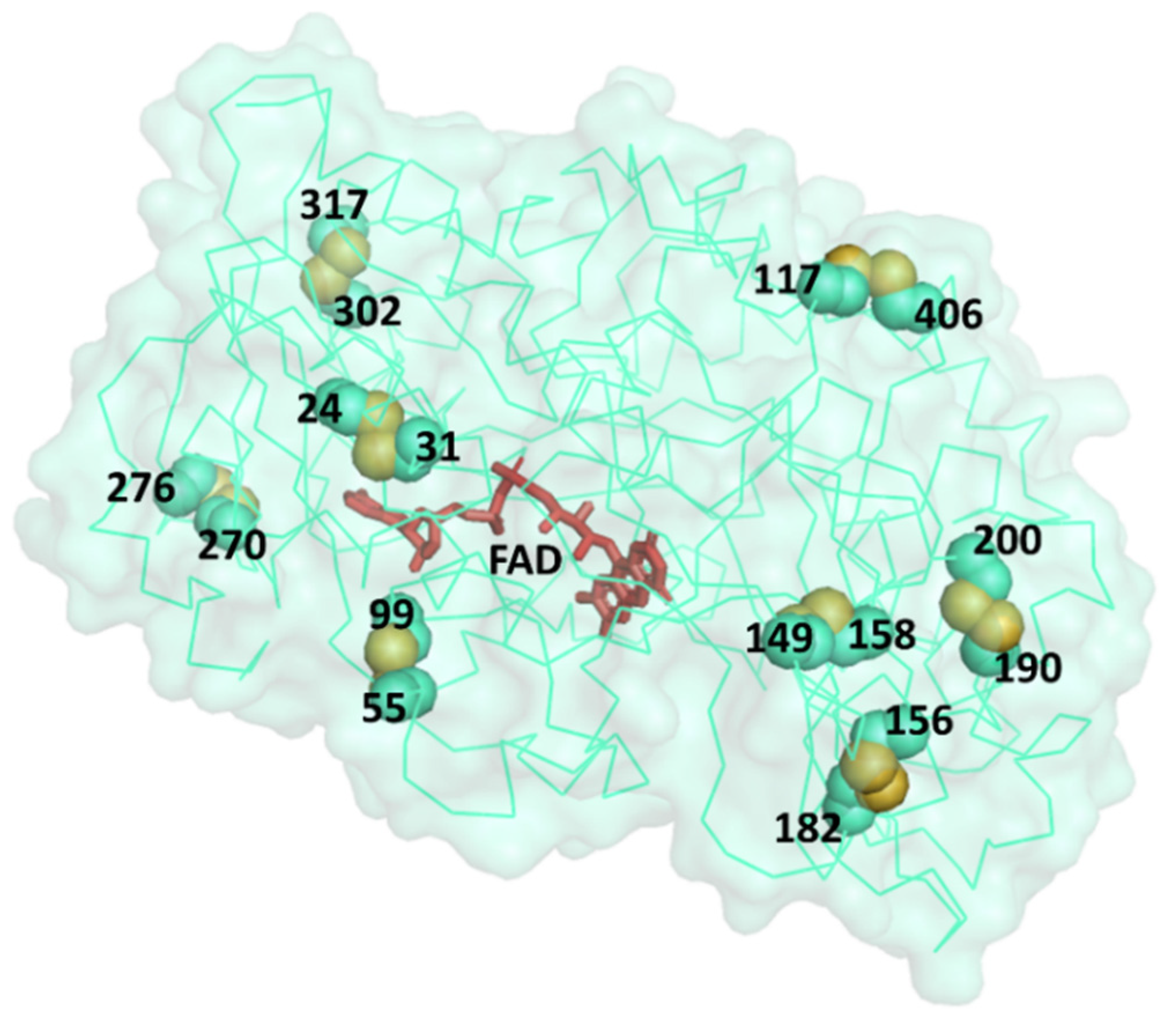
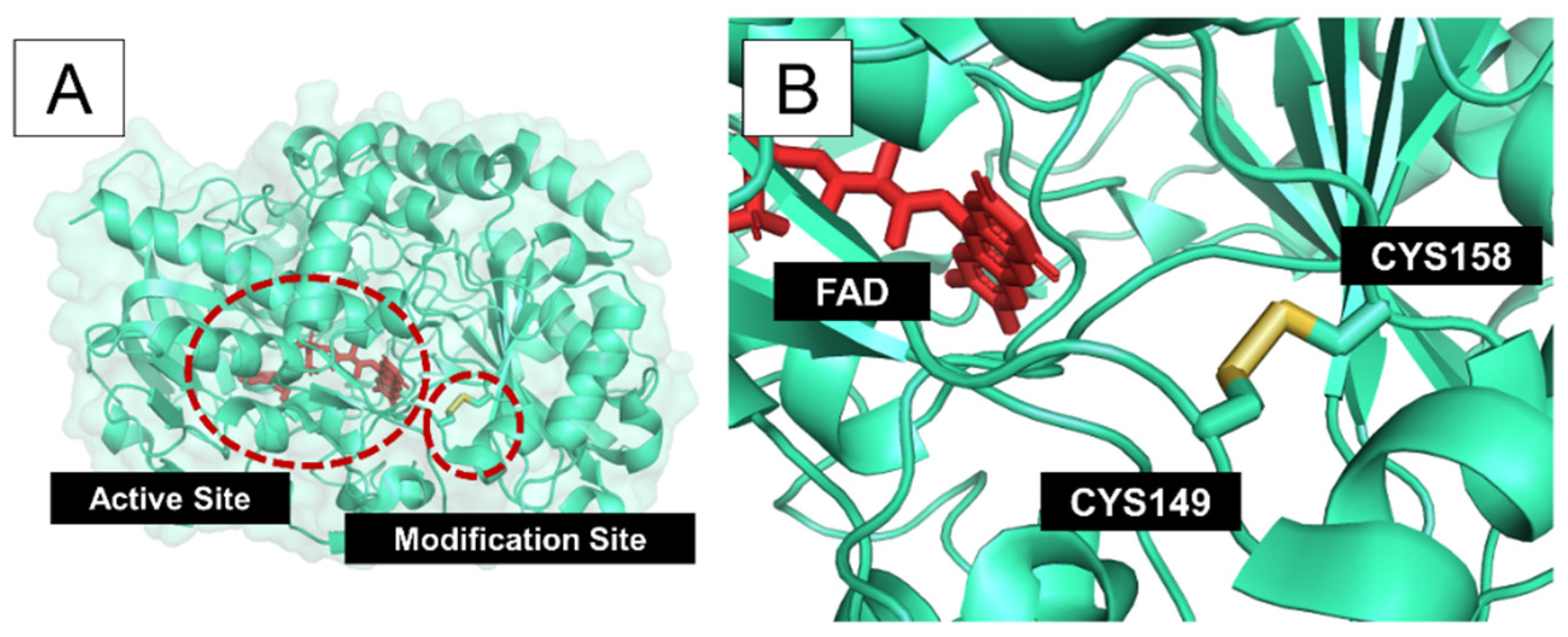
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hocking, A.D. Aspergillus and related teleomorphs. In Food Spoilage Microorganisms; Blackburn, C.D.W., Ed.; Woodhead Publishing, 2006; pp. 451–487. [Google Scholar]
- Dubey, M.K.; Zehra, A.; Aamir, M.; Meena, M.; Ahirwal, L.; Singh, S.; Shukla, S.; Upadhyay, R.S.; Bueno-Mari, R.; Bajpai, V.K. Improvement Strategies, Cost Effective Production, and Potential Applications of Fungal Glucose Oxidase (GOD): Current Updates. Front. Microbiol. 2017, 8, 1032. [Google Scholar] [CrossRef] [Green Version]
- Mano, N. Engineering glucose oxidase for bioelectrochemical applications. Bioelectrochemistry 2019, 128, 218–240. [Google Scholar] [CrossRef]
- Khatami, S.H.; Vakili, O.; Ahmadi, N.; Soltani Fard, E.; Mousavi, P.; Khalvati, B.; Maleksabet, A.; Savardashtaki, A.; Taheri-Anganeh, M.; Movahedpour, A. Glucose oxidase: Applications, sources, and recombinant production. Biotechnol. Appl. Biochem. 2021. [Google Scholar] [CrossRef]
- Jeerapan, I.; Sempionatto, J.R.; Wang, J. On-Body Bioelectronics: Wearable Biofuel Cells for Bioenergy Harvesting and Self-Powered Biosensing. Adv. Funct. Mater. 2020, 30, 1906243. [Google Scholar] [CrossRef]
- Rodrigues, D.; Barbosa, A.I.; Rebelo, R.; Kwon, I.K.; Reis, R.L.; Correlo, V.M. Skin-Integrated Wearable Systems and Implantable Biosensors: A Comprehensive Review. Biosensors 2020, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, J.; Cepra, G. Thermal Stabilization of Enzymes Immobilized within Carbon Paste Electrodes. Anal. Chem. 1997, 69, 3124–3127. [Google Scholar] [CrossRef]
- Yang, D.; Olstad, H.E.; Reyes-De-Corcuera, J.I. Increased thermal stability of a glucose oxidase biosensor under high hydrostatic pressure. Enzym. Microb. Technol. 2020, 134, 109486. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Yang, H.; Shao, Y.; Li, L.; Sun, S.; Wang, L.; Tan, Y.; Xin, Z. Enhancing the activity and thermal stability of a phthalate-degrading hydrolase by random mutagenesis. Ecotoxicol. Environ. Saf. 2021, 209, 111795. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, J.A.; Joo, J.C.; Yoo, Y.J.; Kim, Y.H.; Song, B.K. The development of a thermostable CiP (Coprinus cinereus peroxidase) through in silico design. Biotechnol. Prog. 2010, 26, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef]
- Jacak, R.; Leaver-Fay, A.; Kuhlman, B. Computational protein design with explicit consideration of surface hydrophobic patches. Proteins Struct. Funct. Bioinform. 2012, 80, 825–838. [Google Scholar] [CrossRef] [Green Version]
- Dantas, G.; Kuhlman, B.; Callender, D.; Wong, M.; Baker, D. A Large Scale Test of Computational Protein Design: Folding and Stability of Nine Completely Redesigned Globular Proteins. J. Mol. Biol. 2003, 332, 449–460. [Google Scholar] [CrossRef]
- Broom, A.; Jacobi, Z.; Trainor, K.; Meiering, E.M. Computational tools help improve protein stability but with a solubility tradeoff. J. Biol. Chem. 2017, 292, 14349–14361. [Google Scholar] [CrossRef] [Green Version]
- McRee, D.E. (Ed.) Computational Techniques. In Practical Protein Crystallography, 2nd ed.; Academic Press: San Diego, CA, USA, 1999; p. 91. [Google Scholar]
- Kumar, S.; Nussinov, R. Close-Range Electrostatic Interactions in Proteins. ChemBioChem 2002, 3, 604–617. [Google Scholar] [CrossRef]
- Barik, S. Evolution of Protein Structure and Stability in Global Warming. Int. J. Mol. Sci. 2020, 21, 9662. [Google Scholar] [CrossRef]
- Beeby, M.; O’Connor, B.D.; Ryttersgaard, C.; Boutz, D.R.; Perry, L.J.; Yeates, T.O. The Genomics of Disulfide Bonding and Protein Stabilization in Thermophiles. PLoS Biol. 2005, 3, e309. [Google Scholar] [CrossRef] [Green Version]
- Reed, C.J.; Lewis, H.; Trejo, E.; Winston, V.; Evilia, C. Protein Adaptations in Archaeal Extremophiles. Archaea 2013, 2013, 373275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Yang, T.; Zhou, J.; Xu, M.; Zhang, X.; Rao, Z.; Vieille, C. Elimination of a Free Cysteine by Creation of a Disulfide Bond Increases the Activity and Stability of Candida boidinii Formate Dehydrogenase. Appl. Environ. Microbiol. 2017, 83, e02624-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, L.J.; Wetzel, R. Disulfide Bond Engineered into T4 Lysozyme: Stabilization of the Protein Toward Thermal Inactivation. Science 1984, 226, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.H.; Park, T.Y.; Park, H.J.; Yeon, Y.J.; Yoo, Y.J.; Cha, H.J. Engineering de novo disulfide bond in bacterial α-type carbonic anhydrase for thermostable carbon sequestration. Sci. Rep. 2016, 6, 29322. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins Struct. Funct. Bioinform. 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Hubbard, S.J.; Gross, K.-H.; Argos, P. Intramolecular cavities in globular proteins. Protein Eng. Des. Sel. 1994, 7, 613–626. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Yates, C.M.; Filippis, I.; Kelley, L.A.; Sternberg, M.J.E. SuSPect: Enhanced Prediction of Single Amino Acid Variant (SAV) Phenotype Using Network Features. J. Mol. Biol. 2014, 426, 2692–2701. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef]
- Pazur, J.H. [18] Glucose oxidase from Aspergillus niger. Meth. Enzym. 1966, 9, 82–87. [Google Scholar]
- Jiang, X.; Wang, Y.; Wang, Y.; Huang, H.; Bai, Y.; Su, X.; Zhang, J.; Yao, B.; Tu, T.; Luo, H. Exploiting the activity–stability trade-off of glucose oxidase from Aspergillus niger using a simple approach to calculate thermostability of mutants. Food Chem. 2021, 342, 128270. [Google Scholar] [CrossRef] [PubMed]
- Nagano, N.; Ota, M.; Nishikawa, K. Strong hydrophobic nature of cysteine residues in proteins. FEBS Lett. 1999, 458, 69–71. [Google Scholar] [CrossRef]
- Chen, C.-W.; Lin, M.-H.; Liao, C.-C.; Chang, H.-P.; Chu, Y.-W. iSTable 2.0: Predicting protein thermal stability changes by integrating various characteristic modules. Comput. Struct. Biotechnol. J. 2020, 18, 622–630. [Google Scholar] [CrossRef]
- Sanavia, T.; Birolo, G.; Montanucci, L.; Turina, P.; Capriotti, E.; Fariselli, P. Limitations and challenges in protein stability prediction upon genome variations: Towards future applications in precision medicine. Comput. Struct. Biotechnol. J. 2020, 18, 1968–1979. [Google Scholar] [CrossRef] [PubMed]
- Marabotti, A.; Del Prete, E.; Scafuri, B.; Facchiano, A. Performance of Web tools for predicting changes in protein stability caused by mutations. BMC Bioinform. 2021, 22, 345. [Google Scholar] [CrossRef] [PubMed]
- Hazes, B.; Dijkstra, B.W. Model building of disulfide bonds in proteins with known three-dimensional structure. Protein Eng. Des. Sel. 1988, 2, 119–125. [Google Scholar] [CrossRef]
- Dani, V.S.; Ramakrishnan, C.; Varadarajan, R. MODIP revisited: Re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. Des. Sel. 2003, 16, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Suplatov, D.; Timonina, D.; Sharapova, Y.; Švedas, V. Yosshi: A web-server for disulfide engineering by bioinformatic analysis of diverse protein families. Nucleic Acids Res. 2019, 47, W308–W314. [Google Scholar] [CrossRef]
- Yu, X.-W.; Tan, N.-J.; Xiao, R.; Xu, Y. Engineering a Disulfide Bond in the Lid Hinge Region of Rhizopus chinensis Lipase: Increased Thermostability and Altered Acyl Chain Length Specificity. PLoS ONE 2012, 7, e46388. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.-Y.; Kim, S.; Yun, C.-W.; Choi, Y.-J.; Cho, S.-G. Engineering a de novo internal disulfide bridge to improve the thermal stability of xylanase from Bacillus stearothermophilus No. 236. J. Biotechnol. 2007, 127, 300–309. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| PDB | Resolution (Å) | Residue 1 (PDB) | Residue 2 (PDB) | Cβ–Cβ (Å) | S–S (Å) | α1 (°) | α2 (°) | ΔΔG (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| 3QVP | 1.20 | Ile24 | Gly31 | 4.22 | 2.25 | 110.1 | 99.8 | −2.31 |
| Pro149 | His158 | 3.84 | 2.28 | 111.9 | 103.3 | 0.96 | ||
| Gly270 | Thr276 | 3.68 | 2.19 | 108.5 | 113.7 | −2.59 | ||
| 3QVR | 1.30 | Glu55 | Gly99 | 4.19 | 2.48 | 93.0 | 99.2 | −2.91 |
| Pro149 | His158 | 3.86 | 2.27 | 113.1 | 103.4 | 0.13 | ||
| Gly270 | Thr276 | 3.84 | 2.49 | 114.8 | 104.9 | −2.61 | ||
| Gly302 | Val317 | 3.66 | 2.22 | 110.0 | 93.1 | −1.13 | ||
| 5NIW | 1.80 | Ile24 | Gly31 | 4.21 | 2.61 | 113.7 | 99.2 | −2.33 |
| Pro149 | His158 | 3.83 | 2.31 | 106.1 | 102.5 | 0.14 | ||
| Gly270 | Thr276 | 3.67 | 2.40 | 108.9 | 109.7 | −2.62 | ||
| Gly302 | Val317 | 3.69 | 2.27 | 109.6 | 97.9 | −1.12 | ||
| 5NIT | 1.87 | Ile24 | Gly31 | 4.09 | 2.66 | 108.8 | 95.9 | −2.33 |
| Pro149 | His158 | 3.90 | 2.37 | 109.9 | 102.8 | 0.14 | ||
| Gly270 | Thr276 | 3.62 | 2.48 | 98.2 | 113.2 | −2.62 | ||
| Gly302 | Val317 | 3.69 | 2.14 | 109.6 | 96.2 | −0.96 | ||
| 1CF3 | 1.90 | Pro149 | His158 | 3.96 | 2.26 | 113.7 | 107.9 | 0.99 |
| Gly270 | Thr276 | 3.58 | 2.46 | 99.5 | 111.0 | −2.65 | ||
| 1GAL | 2.30 | Ile24 | Gly31 | 4.21 | 2.36 | 114.4 | 99.4 | −2.33 |
| Ala117 | His406 | 3.88 | 2.66 | 98.7 | 113.6 | 1.37 | ||
| Ala156 | Tyr182 | 3.54 | 1.80 | 103.0 | 96.1 | −2.04 | ||
| Met190 | Thr200 | 4.15 | 2.08 | 105.3 | 93.4 | −3.92 |
| Residue 1 | Conservation Score | Residue 2 | Conservation Score |
|---|---|---|---|
| Ile24 | 0.66 | Gly31 | 0.67 |
| Glu55 | 0.38 | Gly99 | 0.57 |
| Ala117 | 0.43 | His406 | 0.27 |
| Pro149 | 0.29 | His158 | 0.17 |
| Ala156 | 0.29 | Tyr182 | 0.29 |
| Met190 | 0.33 | Thr200 | 0.33 |
| Gly270 | 0.28 | Thr276 | 0.25 |
| Gly302 | 0.72 | Val317 | 0.51 |
Color scale least conserved: 0  1: most conserved 1: most conserved | |||
| Residue 1 | SuSPect Score | Residue 2 | SuSPect Score |
|---|---|---|---|
| Ile24 | 79 | Gly31 | 68 |
| Glu55 | 37 | Gly99 | 36 |
| Ala117 | 14 | His406 | 35 |
| Pro149 | 55 | His158 | 21 |
| Ala156 | 10 | Tyr182 | 31 |
| Met190 | 17 | Thr200 | 24 |
| Gly270 | 24 | Thr276 | 18 |
| Gly302 | 17 | Val317 | 44 |
Color scale predicted neutral: 0  100: predicted deleterious 100: predicted deleterious | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ittisoponpisan, S.; Jeerapan, I. In Silico Analysis of Glucose Oxidase from Aspergillus niger: Potential Cysteine Mutation Sites for Enhancing Protein Stability. Bioengineering 2021, 8, 188. https://doi.org/10.3390/bioengineering8110188
Ittisoponpisan S, Jeerapan I. In Silico Analysis of Glucose Oxidase from Aspergillus niger: Potential Cysteine Mutation Sites for Enhancing Protein Stability. Bioengineering. 2021; 8(11):188. https://doi.org/10.3390/bioengineering8110188
Chicago/Turabian StyleIttisoponpisan, Sirawit, and Itthipon Jeerapan. 2021. "In Silico Analysis of Glucose Oxidase from Aspergillus niger: Potential Cysteine Mutation Sites for Enhancing Protein Stability" Bioengineering 8, no. 11: 188. https://doi.org/10.3390/bioengineering8110188