Comparison of Three Glycoproteomic Methods for the Analysis of the Secretome of CHO Cells Treated with 1,3,4-O-Bu3ManNAc

 ,
, 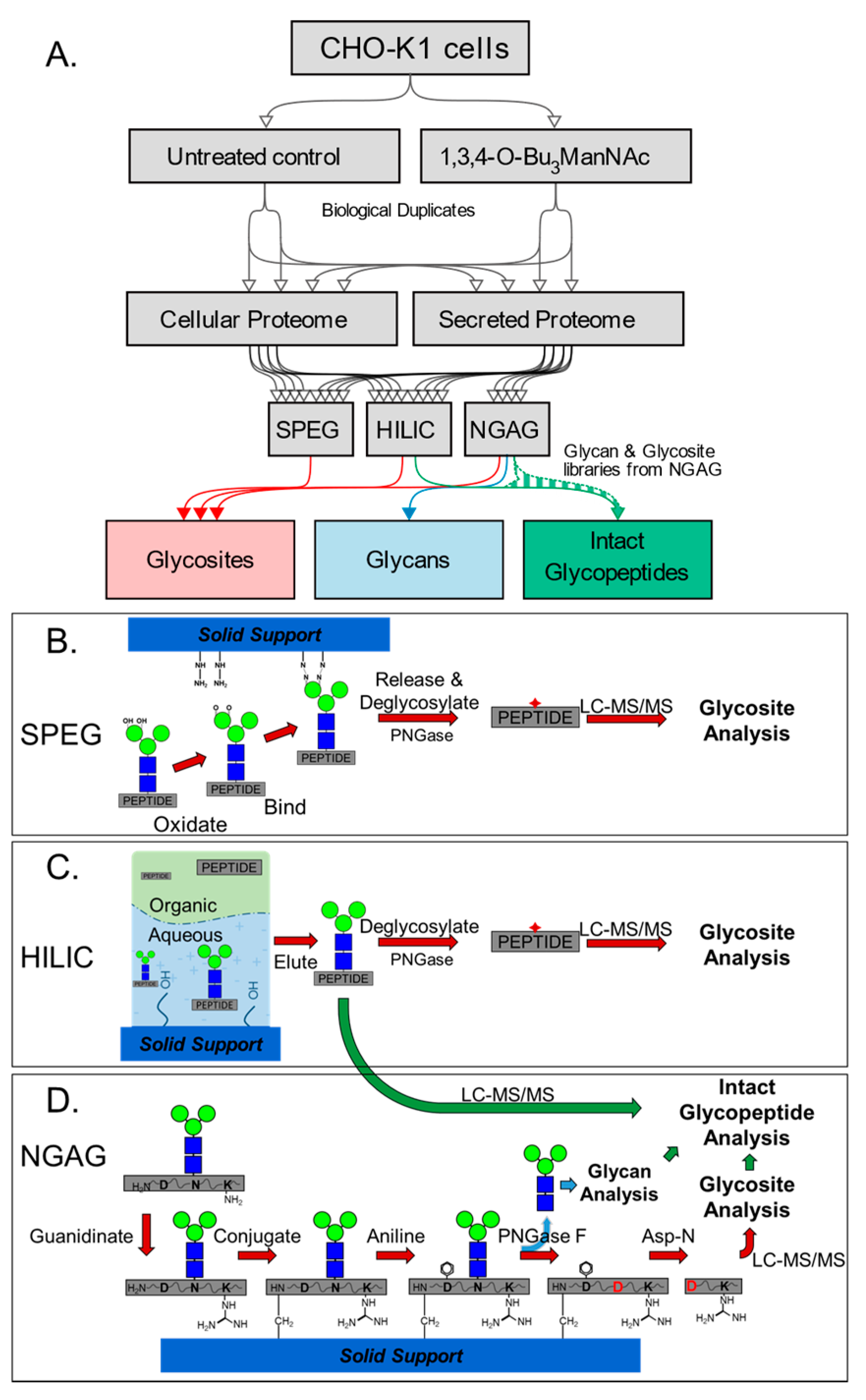{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. CHO Cell Culture and Protein Harvest
2.2. SPEG Enrichment of Deglycosylated Peptides
2.3. HILIC Enrichment of Deglycosylated Peptides and Intact Glycopeptides
2.4. NGAG Enrichment of Deglycosylated Peptides and Glycans
2.5. Mass Spectrometry Analysis of Glycans
2.6. LC-MS/MS Analysis
2.7. Glycosite Data Analysis
2.8. Intact Glycopeptide Data Analysis
3. Results
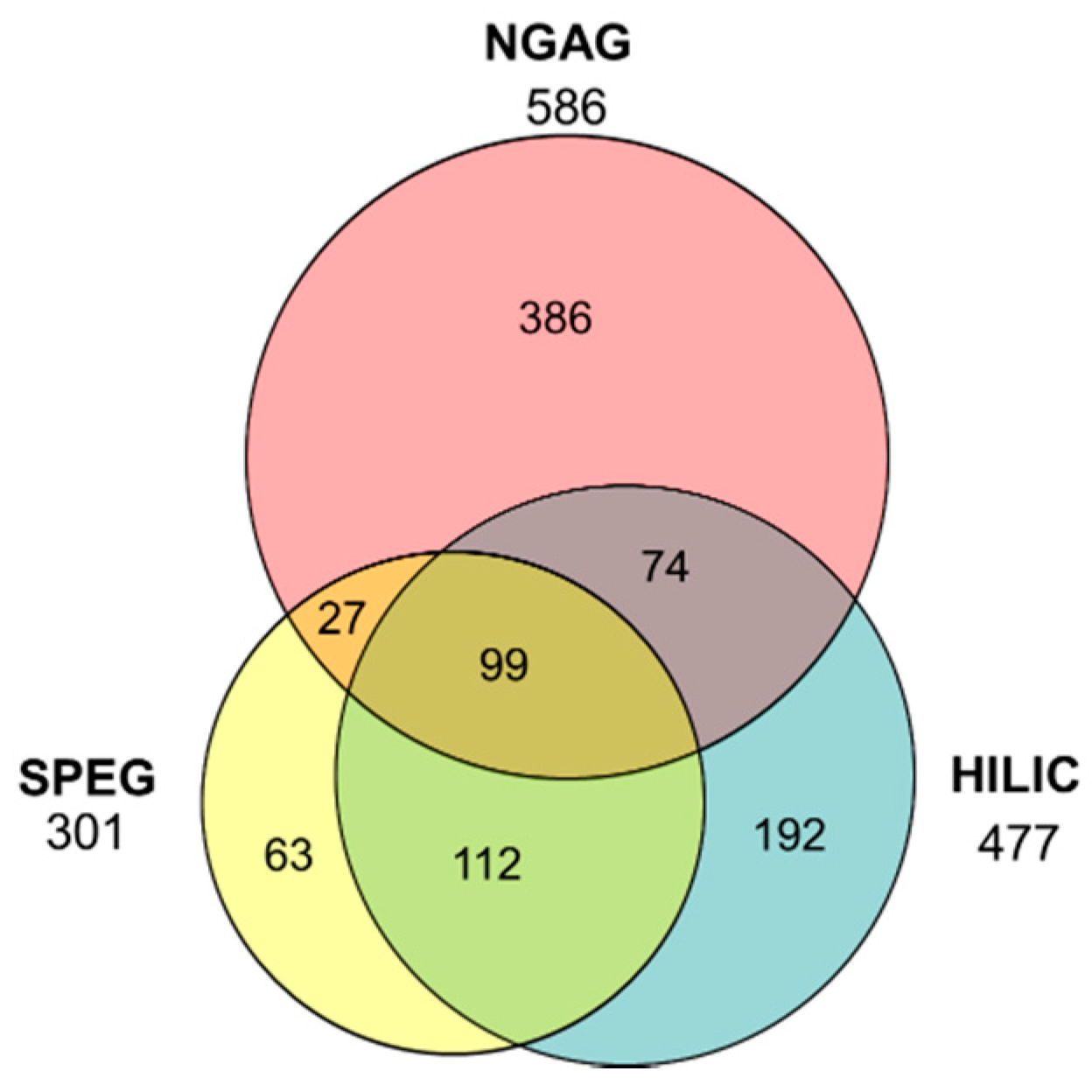
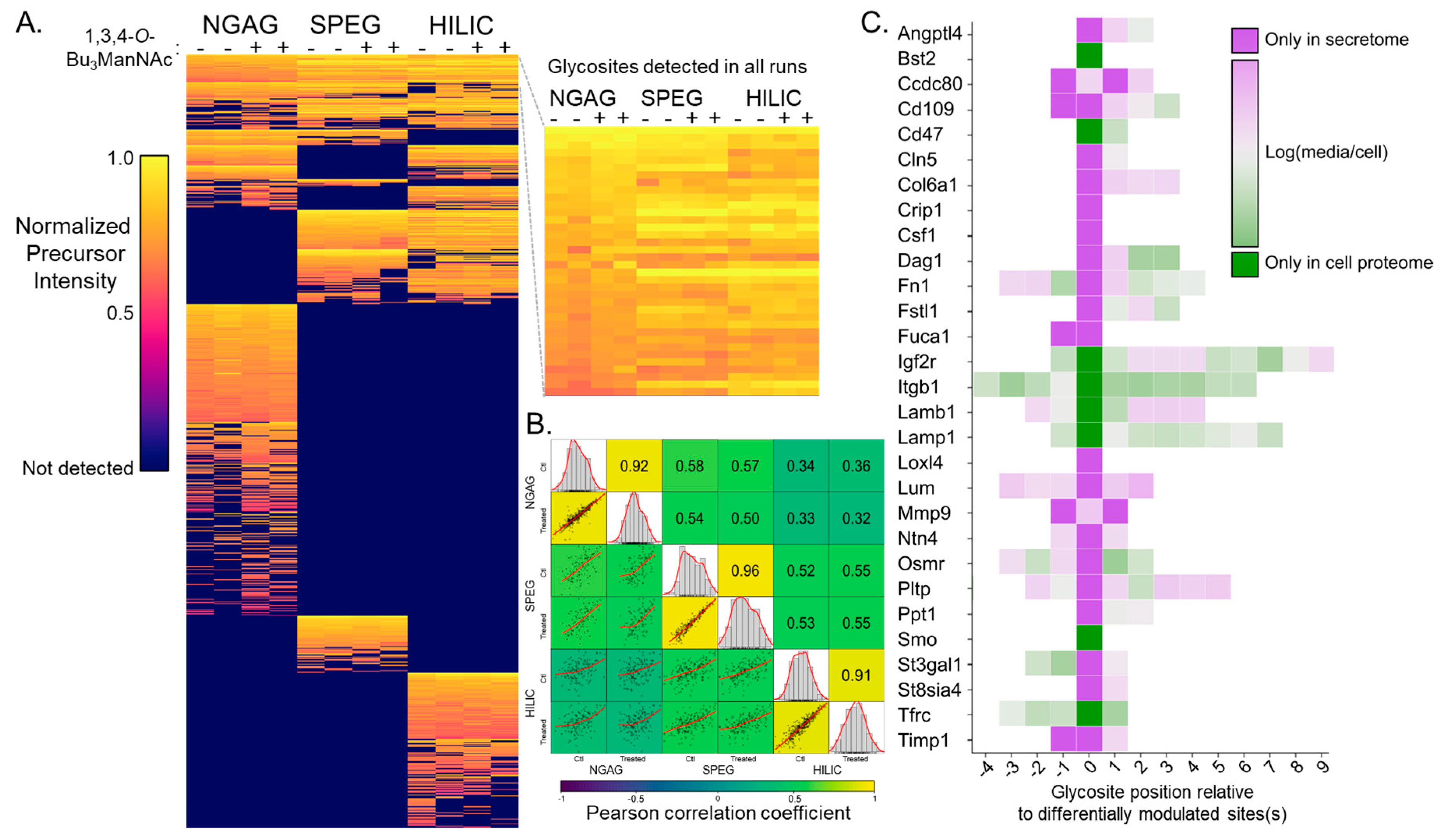
3.1. Quantitative Analysis of CHO N-Glycosites Using Three Glycoproteomic Methods Revealed Complementarity of the NGAG Method
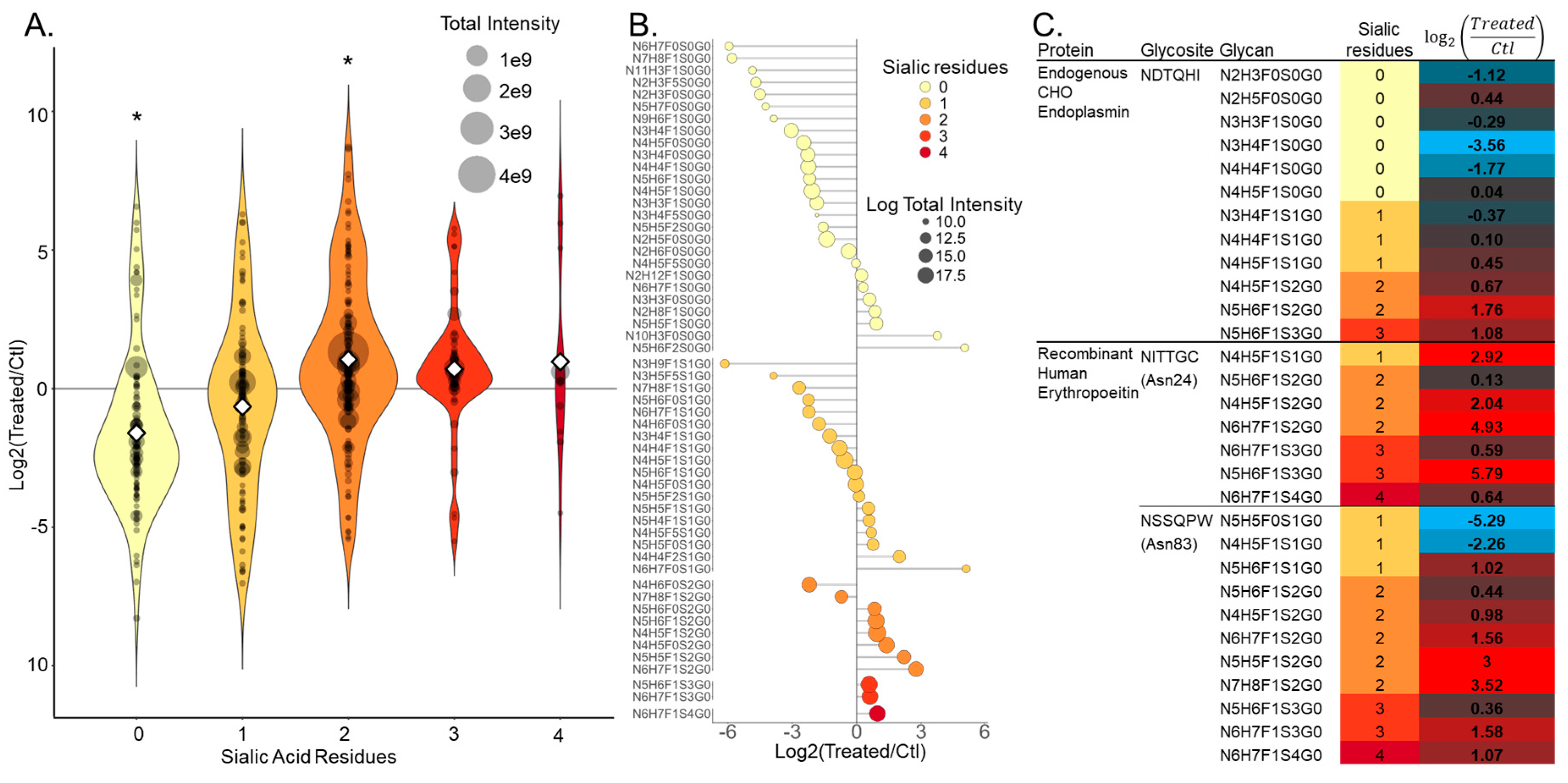
3.2. NGAG Intact Glycopeptide Analysis Revealed Increased Numbers of Secreted Glycosites with Multiple Sialic Acids after Treatment with the Sugar Analog 1,3,4-O-Bu3ManNAc
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, P.M.; Elliott, T.; Cresswell, P.; Wilson, I.A.; Dwek, R.A. Glycosylation and the immune system. Science 2001, 291, 2370–2376. [Google Scholar] [CrossRef]
- Erickson, J.R.; Pereira, L.; Wang, L.; Han, G.; Ferguson, A.; Dao, K.; Copeland, R.J.; Despa, F.; Hart, G.W.; Ripplinger, C.M.; et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013, 502, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv. Cancer Res. 1989, 52, 257–331. [Google Scholar] [PubMed]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, W.; Hu, Y.; Hoti, N.; Liu, Y.; Shah, P.; Sun, S.; Clark, D.; Thomas, S.; Zhang, H. Site-Specific Fucosylation Analysis Identifying Glycoproteins Associated with Aggressive Prostate Cancer Cell Lines Using Tandem Affinity Enrichments of Intact Glycopeptides Followed by Mass Spectrometry. Anal. Chem. 2017, 89, 7623–7630. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.R.; Rodrigues, M.E.; Henriques, M.; Oliveira, R.; Azeredo, J. Glycosylation: Impact, control and improvement during therapeutic protein production. Crit. Rev. Biotechnol. 2014, 34, 281–299. [Google Scholar] [CrossRef]
- Shi, S. Biologics: An update and challenge of their pharmacokinetics. Curr. Drug. Metab. 2014, 15, 271–290. [Google Scholar] [CrossRef]
- Li, H.; d’Anjou, M. Pharmacological significance of glycosylation in therapeutic proteins. Curr. Opin. Biotechnol. 2009, 20, 678–684. [Google Scholar] [CrossRef]
- Baker, K.; Flatman, S.; Birch, J. Product Characterization from Gene to Therapeutic Product. In Medicines from Animal Cell Culture; John Wiley & Sons, Ltd.: Chichester, UK, 2007; pp. 433–442. [Google Scholar]
- Jones, A.J.; Papac, D.I.; Chin, E.H.; Keck, R.; Baughman, S.A.; Lin, Y.S.; Kneer, J.; Battersby, J.E. Selective clearance of glycoforms of a complex glycoprotein pharmaceutical caused by terminal N-acetylglucosamine is similar in humans and cynomolgus monkeys. Glycobiology 2007, 17, 529–540. [Google Scholar] [CrossRef]
- Weigel, P.H.; Yik, J.H. Glycans as endocytosis signals: The cases of the asialoglycoprotein and hyaluronan/chondroitin sulfate receptors. Biochim. Biophys. Acta 2002, 1572, 341–363. [Google Scholar] [CrossRef]
- Walsh, G. Biopharmaceutical benchmarks 2010. Nat. Biotechnol. 2010, 28, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. Mammalian cell protein expression for biopharmaceutical production. Biotechnol. Adv. 2012, 30, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.E.; Liu, X.; Li, Y.; Nagarajan, H.; Yerganian, G.; O’Brien, E.; Bordbar, A.; Roth, A.M.; Rosenbloom, J.; Bian, C.; et al. Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat. Biotechnol. 2013, 31, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Nagarajan, H.; Lewis, N.E.; Pan, S.; Cai, Z.; Liu, X.; Chen, W.; Xie, M.; Wang, W.; Hammond, S.; et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 2011, 29, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Svensson, E.C.; Soreghan, B.; Paulson, J.C. Organization of the beta-galactoside alpha 2,6-sialyltransferase gene. Evidence for the transcriptional regulation of terminal glycosylation. J. Biol. Chem. 1990, 265, 20863–20868. [Google Scholar]
- Butler, M. Optimisation of the cellular metabolism of glycosylation for recombinant proteins produced by Mammalian cell systems. Cytotechnology 2006, 50, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Lin, N.; Mascarenhas, J.; Sealover, N.R.; George, H.J.; Brooks, J.; Kayser, K.J.; Gau, B.; Yasa, I.; Azadi, P.; Archer-Hartmann, S. Chinese hamster ovary (CHO) host cell engineering to increase sialylation of recombinant therapeutic proteins by modulating sialyltransferase expression. Biotechnol. Prog. 2015, 31, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Cha, H.M.; Lim, J.H.; Yeon, J.H.; Hwang, J.M.; Kim, D.I. Co-overexpression of Mgat1 and Mgat4 in CHO cells for production of highly sialylated albumin-erythropoietin. Enzyme Microb. Technol. 2017, 103, 53–58. [Google Scholar] [CrossRef]
- Yin, B.; Gao, Y.; Chung, C.Y.; Yang, S.; Blake, E.; Stuczynski, M.C.; Tang, J.; Kildegaard, H.F.; Andersen, M.R.; Zhang, H.; et al. Glycoengineering of Chinese hamster ovary cells for enhanced erythropoietin N-glycan branching and sialylation. Biotechnol. Bioeng. 2015, 112, 2343–2351. [Google Scholar] [CrossRef]
- Liu, B.; Spearman, M.; Doering, J.; Lattová, E.; Perreault, H.; Butler, M. The availability of glucose to CHO cells affects the intracellular lipid-linked oligosaccharide distribution, site occupancy and the N-glycosylation profile of a monoclonal antibody. J. Biotechnol. 2014, 170, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryczko, M.C.; Pawling, J.; Chen, R.; Abdel Rahman, A.M.; Yau, K.; Copeland, J.K.; Zhang, C.; Surendra, A.; Guttman, D.S.; Figeys, D.; et al. Metabolic Reprogramming by Hexosamine Biosynthetic and Golgi N-Glycan Branching Pathways. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Crowell, C.K.; Grampp, G.E.; Rogers, G.N.; Miller, J.; Scheinman, R.I. Amino acid and manganese supplementation modulates the glycosylation state of erythropoietin in a CHO culture system. Biotechnol. Bioeng. 2007, 96, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wang, D.I.C. Improvement of interferon-γ sialylation in Chinese hamster ovary cell culture by feeding ofN-acetylmannosamine. Biotechnol. Bioeng. 1998, 58, 642–648. [Google Scholar] [CrossRef]
- Sung, Y.H.; Song, Y.J.; Lim, S.W.; Chung, J.Y.; Lee, G.M. Effect of sodium butyrate on the production, heterogeneity and biological activity of human thrombopoietin by recombinant Chinese hamster ovary cells. J. Biotechnol. 2004, 112, 323–335. [Google Scholar] [CrossRef]
- Wang, Q.; Chung, C.Y.; Yang, W.; Yang, G.; Chough, S.; Chen, Y.; Yin, B.; Bhattacharya, R.; Hu, Y.; Saeui, C.T.; et al. Combining Butyrated ManNAc with Glycoengineered CHO Cells Improves EPO Glycan Quality and Production. Biotechnol. J. 2019, 14, e1800186. [Google Scholar] [CrossRef]
- Yin, B.; Wang, Q.; Chung, C.Y.; Bhattacharya, R.; Ren, X.; Tang, J.; Yarema, K.J.; Betenbaugh, M.J. A novel sugar analog enhances sialic acid production and biotherapeutic sialylation in CHO cells. Biotechnol. Bioeng. 2017, 114, 1899–1902. [Google Scholar] [CrossRef]
- Almaraz, R.T.; Tian, Y.; Bhattarcharya, R.; Tan, E.; Chen, S.H.; Dallas, M.R.; Chen, L.; Zhang, Z.; Zhang, H.; Konstantopoulos, K.; et al. Metabolic flux increases glycoprotein sialylation: Implications for cell adhesion and cancer metastasis. Mol. Cell Proteom. 2012, 11. [Google Scholar] [CrossRef] [Green Version]
- Saeui, C.T.; Cho, K.C.; Dharmarha, V.; Nairn, A.V.; Galizzi, M.; Shah, S.R.; Gowda, P.; Park, M.; Austin, M.; Clarke, A.; et al. Cell Line-, Protein-, and Sialoglycosite-Specific Control of Flux-Based Sialylation in Human Breast Cells: Implications for Cancer Progression. Front. Chem. 2020, 8, 13. [Google Scholar] [CrossRef]
- Zielinska, D.F.; Gnad, F.; Wisniewski, J.R.; Mann, M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 2010, 141, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; LaRoche, T.; Hamelinck, D.; Bergsma, D.; Brenner, D.; Simeone, D.; Brand, R.E.; Haab, B.B. Multiplexed analysis of glycan variation on native proteins captured by antibody microarrays. Nat. Methods 2007, 4, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, H.; Tang, H.; Yang, P. Endoglycosidase-mediated incorporation of 18O into glycans for relative glycan quantitation. Anal. Chem. 2011, 83, 4975–4981. [Google Scholar] [CrossRef]
- Shah, P.; Yang, S.; Sun, S.; Aiyetan, P.; Yarema, K.J.; Zhang, H. Mass spectrometric analysis of sialylated glycans with use of solid-phase labeling of sialic acids. Anal. Chem. 2013, 85, 3606–3613. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, X.J.; Martin, D.B.; Aebersold, R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003, 21, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.L.; Toonstra, C.; Zhang, H. Chemical Strategies to Glycoprotein Analysis. In Mass Spectrometry-Based Chemical Proteomics; John Wiley & Sons, Inc.: Chichester, UK, 2019; pp. 293–316. [Google Scholar]
- Sun, S.; Hu, Y.; Ao, M.; Shah, P.; Chen, J.; Yang, W.; Jia, X.; Tian, Y.; Thomas, S.; Zhang, H. N-GlycositeAtlas: A database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clin. Proteom. 2019, 16, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Gaiki, S. Retention and selectivity of stationary phases for hydrophilic interaction chromatography. J. Chromatogr. A 2011, 1218, 5920–5938. [Google Scholar] [CrossRef]
- Yang, G.; Hu, Y.; Sun, S.; Ouyang, C.; Yang, W.; Wang, Q.; Betenbaugh, M.; Zhang, H. Comprehensive Glycoproteomic Analysis of Chinese Hamster Ovary Cells. Anal. Chem. 2018, 90, 14294–14302. [Google Scholar] [CrossRef]
- Wu, S.W.; Liang, S.Y.; Pu, T.H.; Chang, F.Y.; Khoo, K.H. Sweet-Heart—An integrated suite of enabling computational tools for automated MS2/MS3 sequencing and identification of glycopeptides. J. Proteom. 2013, 84, 1–16. [Google Scholar] [CrossRef]
- Sun, S.; Shah, P.; Eshghi, S.T.; Yang, W.; Trikannad, N.; Yang, S.; Chen, L.; Aiyetan, P.; Hoti, N.; Zhang, Z.; et al. Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat. Biotechnol. 2016, 34, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Shah, P.; Clark, D.J.; Ao, M.; Zhang, H. Reanalysis of Global Proteomic and Phosphoproteomic Data Identified a Large Number of Glycopeptides. Anal. Chem. 2018, 90, 8065–8071. [Google Scholar] [CrossRef]
- Toghi Eshghi, S.; Shah, P.; Yang, W.; Li, X.; Zhang, H. GPQuest: A Spectral Library Matching Algorithm for Site-Specific Assignment of Tandem Mass Spectra to Intact N-glycopeptides. Anal. Chem. 2015, 87, 5181–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aich, U.; Campbell, C.T.; Elmouelhi, N.; Weier, C.A.; Sampathkumar, S.G.; Choi, S.S.; Yarema, K.J. Regioisomeric SCFA attachment to hexosamines separates metabolic flux from cytotoxicity and MUC1 suppression. ACS Chem. Biol. 2008, 3, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shah, P.; Hu, Y.; Eshghi, S.T.; Sun, S.; Liu, Y.; Zhang, H. Comparison of Enrichment Methods for Intact N- and O-Linked Glycopeptides Using Strong Anion Exchange and Hydrophilic Interaction Liquid Chromatography. Anal. Chem. 2017, 89, 11193–11197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von der Lieth, C.W.; Freire, A.A.; Blank, D.; Campbell, M.P.; Ceroni, A.; Damerell, D.R.; Dell, A.; Dwek, R.A.; Ernst, B.; Fogh, R.; et al. EUROCarbDB: An open-access platform for glycoinformatics. Glycobiology 2010, 21, 493–502. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Michalski, A.; Damoc, E.; Hauschild, J.P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell Proteom. 2011, 10, M111.011015. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhu, G.; Dovichi, N.J. Comparison of the LTQ-Orbitrap Velos and the Q-Exactive for proteomic analysis of 1–1000 ng RAW 264.7 cell lysate digests. Rapid Commun. Mass Spectrom. 2013, 27, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Du, J.; Che, P.L.; Meledeo, M.A.; Yarema, K.J. Hexosamine analogs: From metabolic glycoengineering to drug discovery. Curr. Opin. Chem. Biol. 2009, 13, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Mathew, M.P.; Tan, E.; Shah, S.; Bhattacharya, R.; Meledeo, M.A.; Huang, J.; Espinoza, F.A.; Yarema, K.J. Extracellular and intracellular esterase processing of SCFA-hexosamine analogs: Implications for metabolic glycoengineering and drug delivery. Bioorg. Med. Chem. Lett. 2012, 22, 6929–6933. [Google Scholar] [CrossRef] [Green Version]
- Tsiatsiani, L.; Heck, A.J. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef]
- Vincent, D.; Ezernieks, V.; Rochfort, S.; Spangenberg, G. A Multiple Protease Strategy to Optimise the Shotgun Proteomics of Mature Medicinal Cannabis Buds. Int. J. Mol. Sci. 2019, 20, 5630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaney, D.L.; Wenger, C.D.; Coon, J.J. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 2010, 9, 1323–1329. [Google Scholar] [CrossRef] [Green Version]
- Dau, T.; Bartolomucci, G.; Rappsilber, J. Proteomics Using Protease Alternatives to Trypsin Benefits from Sequential Digestion with Trypsin. Anal. Chem. 2020, 92, 9523–9527. [Google Scholar] [CrossRef] [PubMed]
- Brune, D.; Andrade-Navarro, M.A.; Mier, P. Proteome-wide comparison between the amino acid composition of domains and linkers. BMC Res. Notes 2018, 11, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Niwa, Y.; Suzuki, T.; Uematsu, S.; Dohmae, N.; Simizu, S. N-glycosylation is required for secretion and enzymatic activity of human hyaluronidase1. FEBS Open Bio 2014, 4, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Yu, X.W.; Zheng, H.; Sha, C.; Zhao, C.; Qian, M.; Xu, Y. Role of N-linked glycosylation in the secretion and enzymatic properties of Rhizopus chinensis lipase expressed in Pichia pastoris. Microb. Cell Fact. 2015, 14, 40. [Google Scholar] [CrossRef] [Green Version]
- Sagt, C.M.; Kleizen, B.; Verwaal, R.; de Jong, M.D.; Muller, W.H.; Smits, A.; Visser, C.; Boonstra, J.; Verkleij, A.J.; Verrips, C.T. Introduction of an N-glycosylation site increases secretion of heterologous proteins in yeasts. Appl. Environ. Microbiol. 2000, 66, 4940–4944. [Google Scholar] [CrossRef] [Green Version]
- Mathew, M.P.; Tan, E.; Saeui, C.T.; Bovonratwet, P.; Sklar, S.; Bhattacharya, R.; Yarema, K.J. Metabolic flux-driven sialylation alters internalization, recycling, and drug sensitivity of the epidermal growth factor receptor (EGFR) in SW1990 pancreatic cancer cells. Oncotarget 2016, 7, 66491–66511. [Google Scholar] [CrossRef]
- Gong, B.; Burnina, I.; Stadheim, T.A.; Li, H. Glycosylation characterization of recombinant human erythropoietin produced in glycoengineered Pichia pastoris by mass spectrometry. J. Mass Spectrom. 2013, 48, 1308–1317. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Ji, Y.; Silbern, I.; Doebele, C.; Ninov, M.; Lenz, C.; Oellerich, T.; Pan, K.-T.; Urlaub, H. A streamlined pipeline for multiplexed quantitative site-specific N-glycoproteomics. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mertz, J.L.; Sun, S.; Yin, B.; Hu, Y.; Bhattacharya, R.; Bettenbaugh, M.J.; Yarema, K.J.; Zhang, H. Comparison of Three Glycoproteomic Methods for the Analysis of the Secretome of CHO Cells Treated with 1,3,4-O-Bu3ManNAc. Bioengineering 2020, 7, 144. https://doi.org/10.3390/bioengineering7040144
Mertz JL, Sun S, Yin B, Hu Y, Bhattacharya R, Bettenbaugh MJ, Yarema KJ, Zhang H. Comparison of Three Glycoproteomic Methods for the Analysis of the Secretome of CHO Cells Treated with 1,3,4-O-Bu3ManNAc. Bioengineering. 2020; 7(4):144. https://doi.org/10.3390/bioengineering7040144
Chicago/Turabian StyleMertz, Joseph L., Shisheng Sun, Bojiao Yin, Yingwei Hu, Rahul Bhattacharya, Michael J. Bettenbaugh, Kevin J. Yarema, and Hui Zhang. 2020. "Comparison of Three Glycoproteomic Methods for the Analysis of the Secretome of CHO Cells Treated with 1,3,4-O-Bu3ManNAc" Bioengineering 7, no. 4: 144. https://doi.org/10.3390/bioengineering7040144