Flax Biomass Conversion via Controlled Oxidation: Facile Tuning of Physicochemical Properties


Abstract
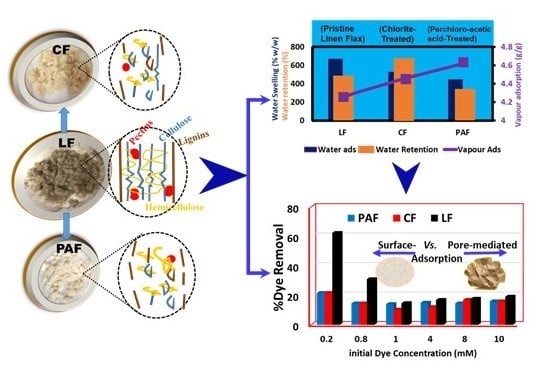
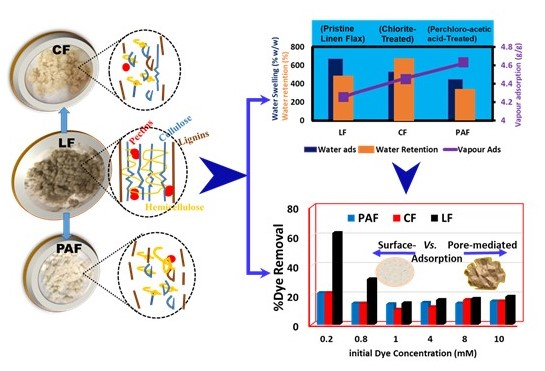
:
1. Introduction
2. Materials and Methods
2.1. Pretreatment of Raw Linen Fiber
2.2. Characterization Methods of Raw Linen Fiber and Its Treated Forms
2.2.1. Compositional Analysis
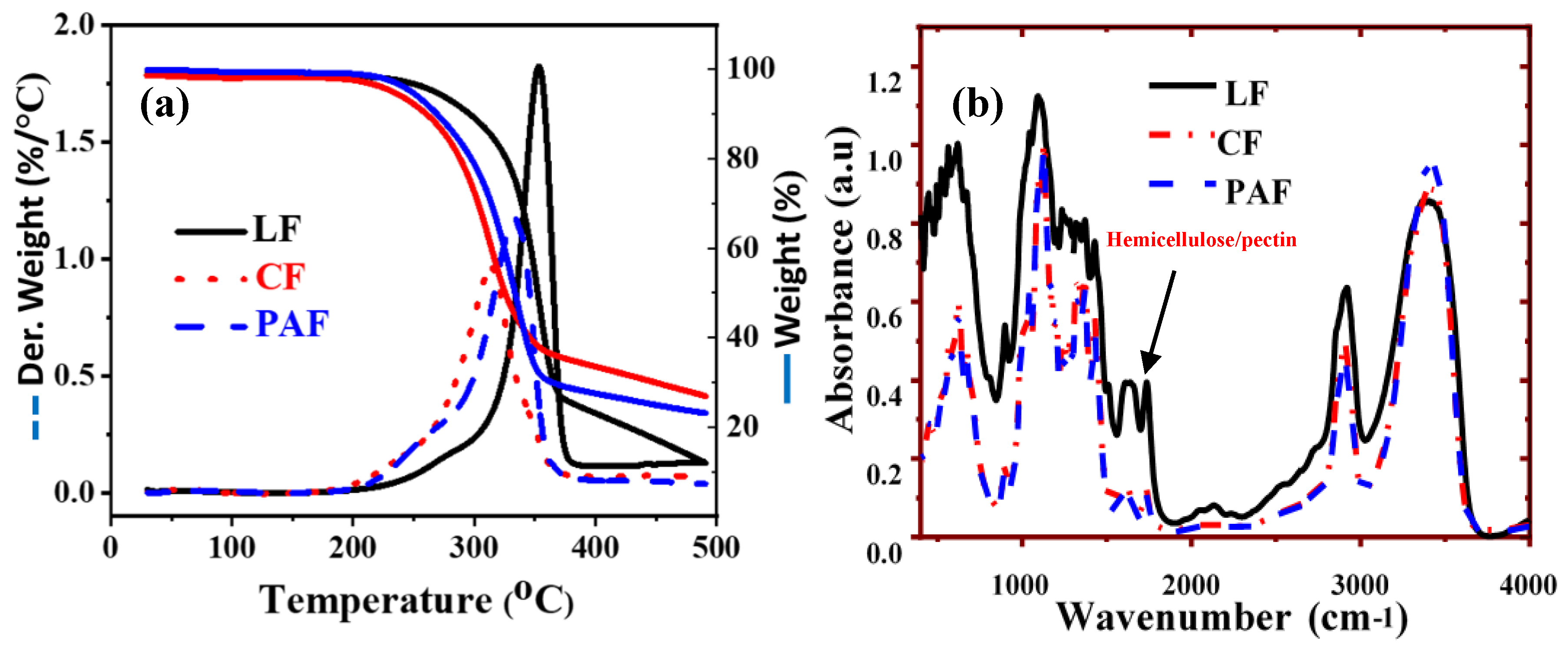
2.2.2. FT-IR, TGA, Nitrogen Gas Adsorption, SEM and TEM
2.2.3. Raman Spectroscopy
2.2.4. Particle Size Distribution and Zeta Potential
2.2.5. X-ray Photoelectron Spectroscopy
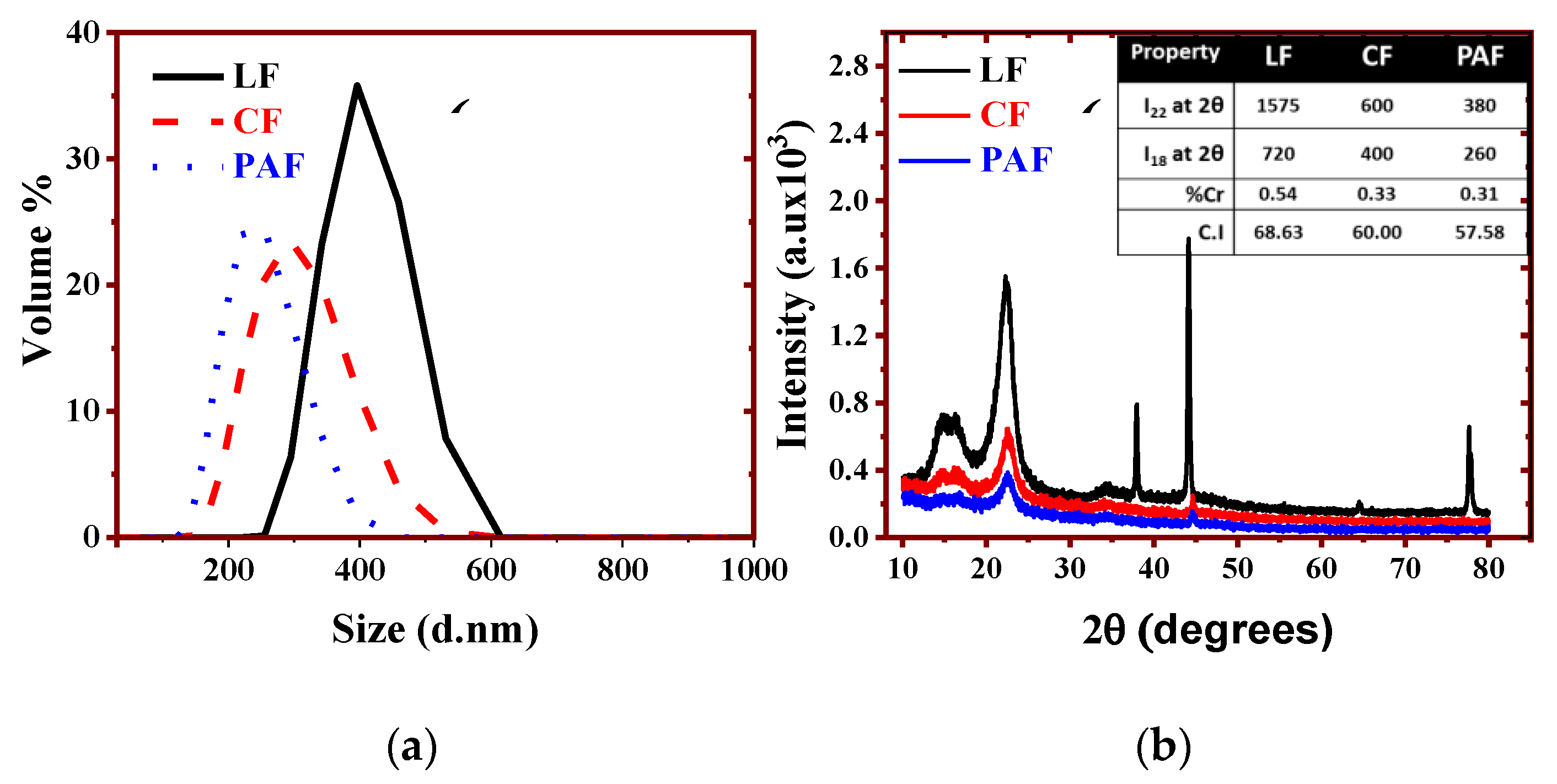
2.2.6. X-ray Diffraction
2.2.7. Differential Scanning Calorimetry
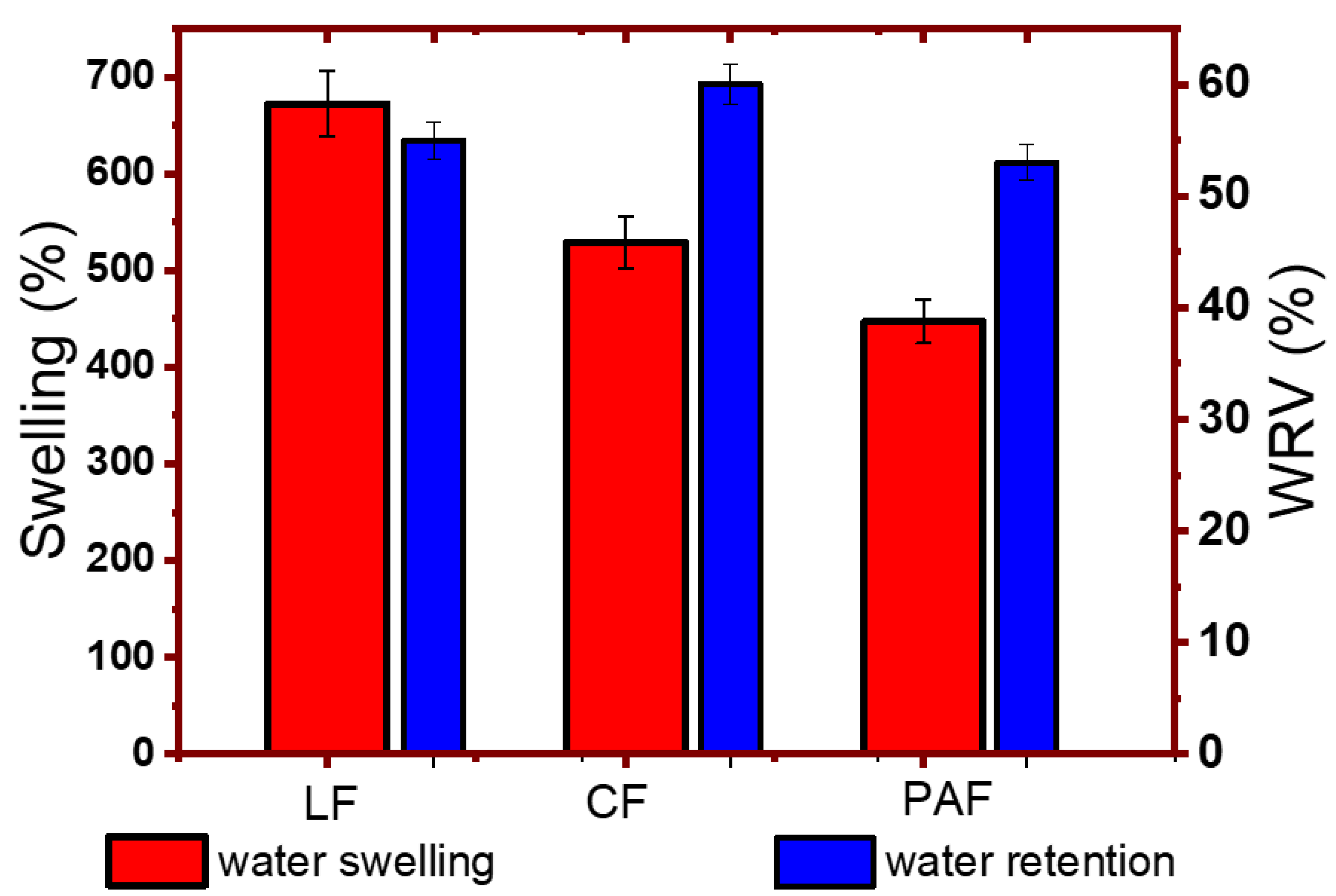
2.2.8. Gravimetric Water Swelling Tests
2.2.9. Water Retention Value (WRV)
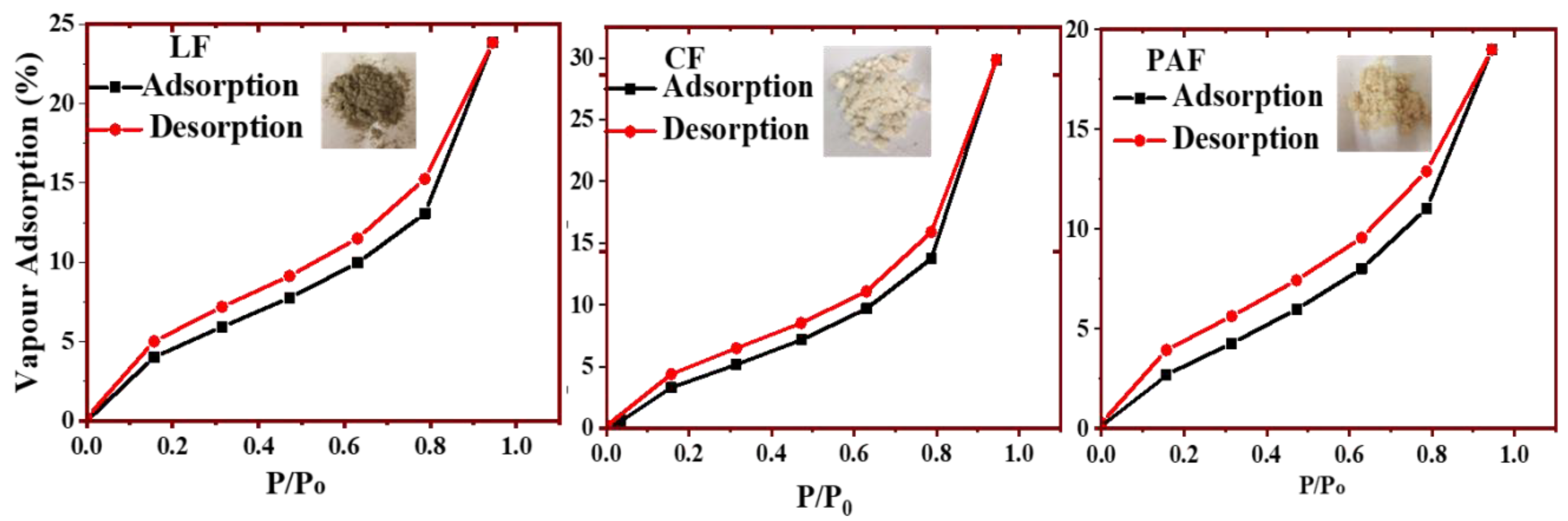
2.2.10. Water Vapour Adsorption Isotherms
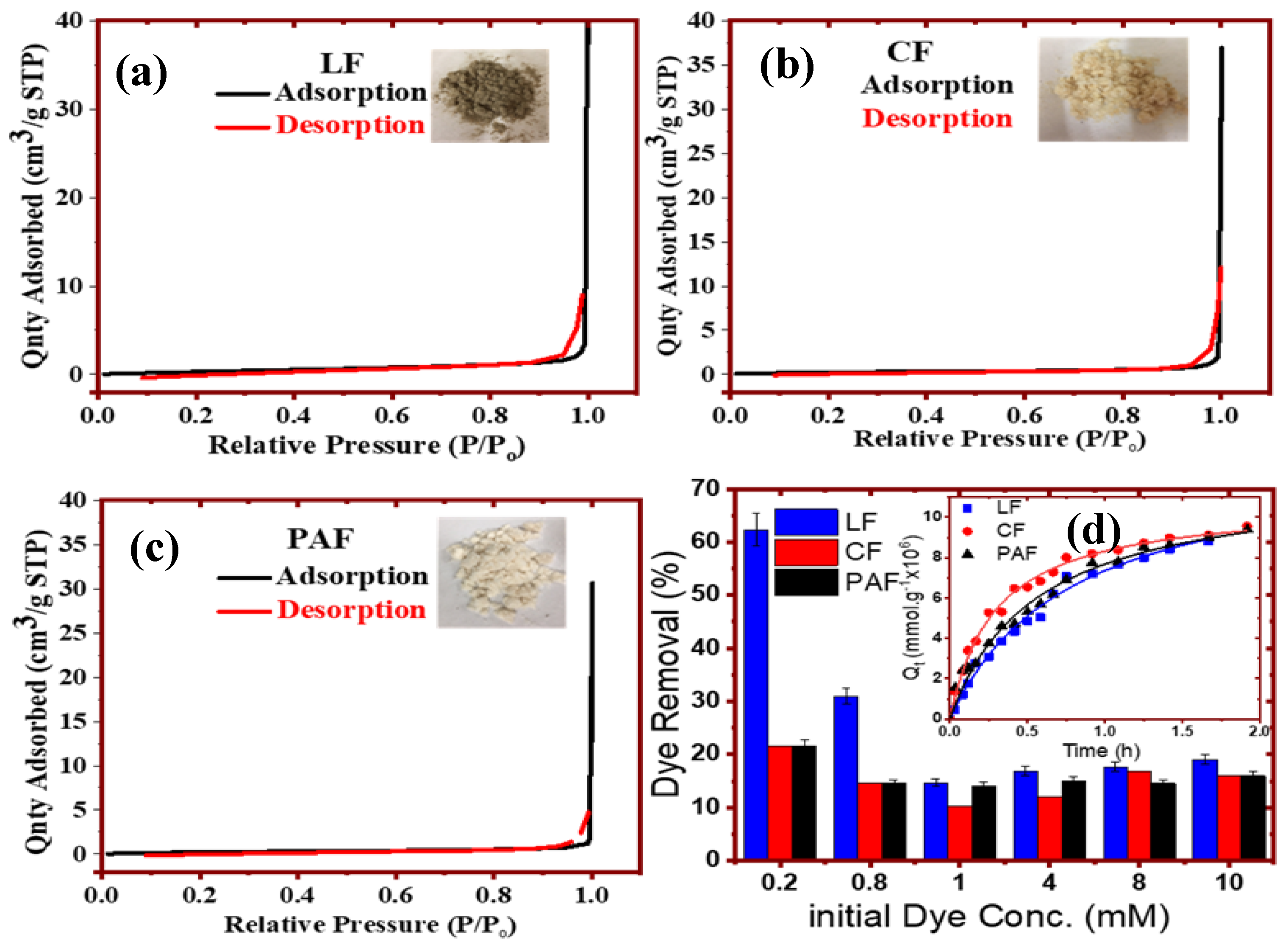
2.2.11. Dye-Probe Kinetic Uptake Studies
3. Results
3.1. Characterization of Linen Fiber and Treated Forms
3.1.1. Compositional Analysis of Pristine and Treated Linen Fiber
3.1.2. Particle Size Distribution (PSD) and Powder X-ray Diffraction (PXRD)
3.1.3. Thermal Gravimetric Analysis (TGA) and FT-IR Spectroscopy
3.2. Textural and Surface Properties
3.2.1. Scanning (SEM) and Transmission Electron Microscopy (TEM)
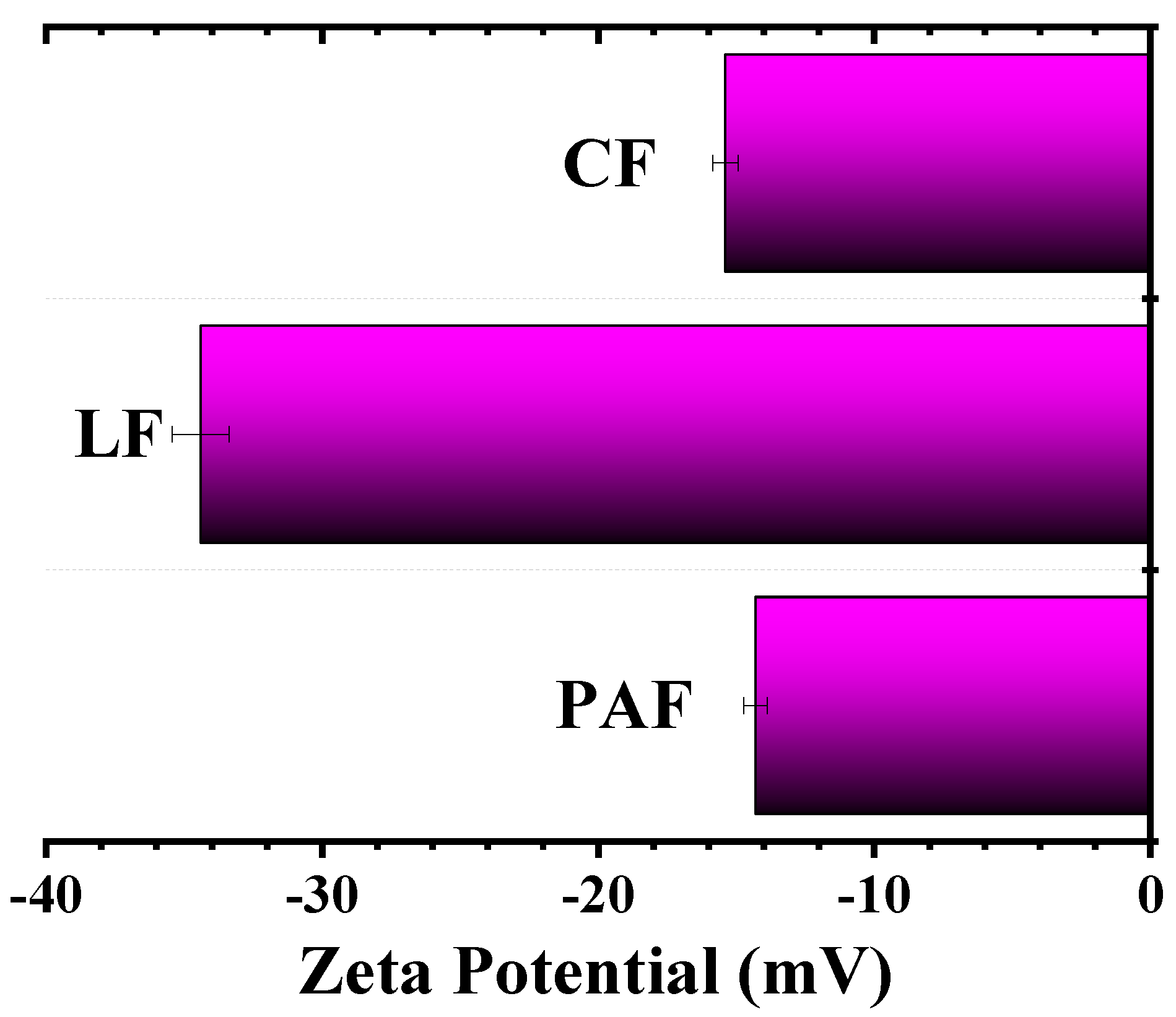
3.2.2. Zeta Potential (ξ) Results
3.2.3. X-ray Photoelectron Spectroscopy (XPS)
4. Discussion
4.1. Hydration Properties of Pristine and Treated Linen Fibers
4.1.1. Water Vapour Uptake by Linen Fiber and Its Modified Forms
4.1.2. Gravimetric Water Swelling and Water Retention Value of Linen Fibers
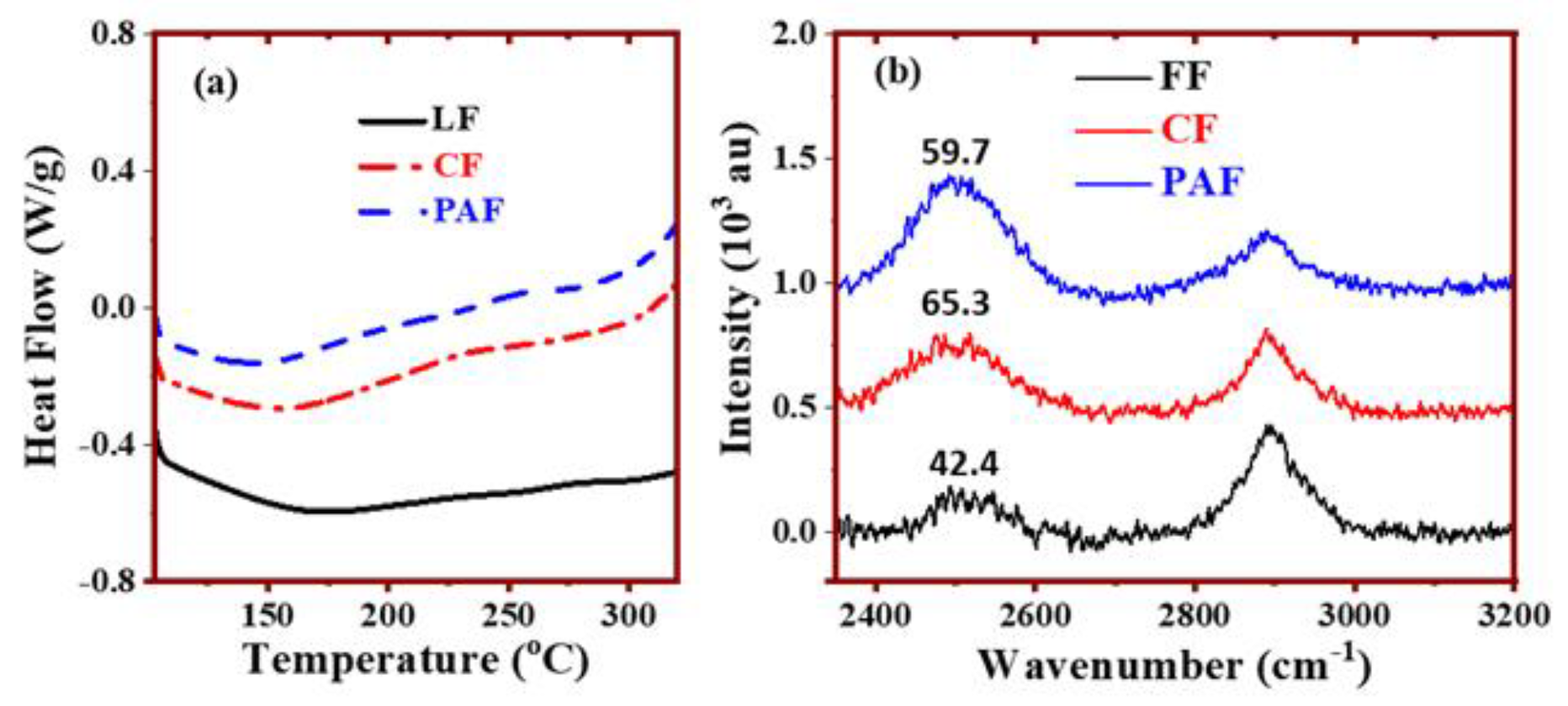
4.2. Thermal Analysis and Raman Spectroscopy of Hydrated Biomaterials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gupta, G.K.; De, S.; Franco, A.; Balu, A.M.; Luque, R. Sustainable Biomaterials: Current Trends, Challenges and Applications. Molecules 2016, 21, 48. [Google Scholar] [CrossRef] [PubMed]
- Irvine, R.B.; McConnell, J.; Lafond, G.P.; May, W.E.; Hultgreen, G.; Ulrich, A.; Stonehouse, K.; Chalmers, S.; Stevenson, F.C. Impact of production practices on fiber yield of oilseed flax under Canadian prairie conditions. Can. J. Plant. Sci. 2010, 90, 61–70. [Google Scholar] [CrossRef]
- Akin, D.; Foulk, J.; Dodd, R.; McAlister, D. Enzyme-retting of flax and characteristics of processed fibers. J. Biotechnol. 2001, 89, 193–203. [Google Scholar] [CrossRef]
- De Prez, J.; Van Vuure, A.W.; Ivens, J.; Aerts, G.; Van de Voorde, I. Enzymatic treatment of flax for use in composites. Biotechnol. Rep. 2018, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Chouw, N.; Jayaraman, M. Flax fibre and its composites—A review. Compos. Part. B Eng. 2014, 56, 296–317. [Google Scholar] [CrossRef]
- Roach, M.J.; Mokshina, N.Y.; Badhan, A.; Snegireva, A.V.; Hobson, N.; Deyholos, M.K.; Gorshkova, T.A. Development of Cellulosic Secondary Walls in Flax Fibers Requires b-Galactosidase. Plant. Physiol. 2011, 156, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Célino, A.; Fréour, S.; Jacquemin, F.; Casari, P. The hygroscopic behavior of plant fibers: A review. Front. Chem. 2014, 1, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Voragen, A.G.J.; Coenen, G.-J.; Verhoef, R.P.; Schols, H.A. Pectin, a versatile polysaccharide presents in plant cell walls. Struct. Chem. 2009, 20, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Berthold, J.; Olsson, R.J.O.; Salmén, L. Water Sorption to Hydroxyl and Carboxylic Acid Groups in Carboxymethylcellulose (CMC) Studied with NIR-spectroscopy. Cellulose 1998, 5, 281–298. [Google Scholar] [CrossRef]
- Konduri, M.; Kong, F.; Fatehi, P. Production of carboxymethylated lignin and its application as a dispersant. Eur. Polym. J. 2015, 70, 371–383. [Google Scholar] [CrossRef]
- Li, X.; Tabil, L.G.; Panigrahi, S. Chemical treatments of natural fiber for use in natural fiber-reinforced composites: A review. J. Polym. Environ. 2007, 15, 25–33. [Google Scholar] [CrossRef]
- Kumar, P.; Saraswat, C.; Mishra, B.K.; Avtar, R.; Patel, H.; Patel, A.; Sharma, T.; Patel, R. Batch technique to evaluate the efficiency of different natural adsorbents for defluoridation from groundwater. Appl. Water Sci. 2017, 7, 2597–2606. [Google Scholar] [CrossRef] [Green Version]
- Karoyo, A.H.; Dehabadi, L.; Alabi, W.; Simonson, C.J.; Wilson, L.D. Hydration and Sorption Properties of Raw and Milled Flax Fibers. ACS Omega 2020, 5, 6113–6121. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.M.; Wang, H.; Lau, K.T.; Cardona, F. Chemical treatments on plant-based natural fibre reinforced polymer composites: An overview. Compos. Part. B Eng. 2012, 43, 2883–2892. [Google Scholar] [CrossRef]
- Ahmad, R.; Hamid, R.; Osman, S.A. Physical and Chemical Modifications of Plant Fibres for Reinforcement in Cementitious Composites. Adv. Civ. Eng. 2019, 2019, 1–19. [Google Scholar] [CrossRef]
- Alix, S.; Marais, S.; Morvan, M.; Lebrun, L. Biocomposite materials from flax plants: Preparation and properties. Compos. Part A Appl. Sci. Manuf. 2008, 39, 1793–1801. [Google Scholar] [CrossRef]
- Dicker, M.; Duckworth, P.F.; Baker, A.; Francois, G. Green composites: A review of material attributes and complementary applications. Compos. Part A Appl. Sci. Manuf. 2014, 56, 280–289. [Google Scholar] [CrossRef]
- Wang, B.; Panjgrahi, S.; Tabil, L.G.; Crerar, W. Modification of flax fiber by chemical treatment. In Proceedings of the CSAE/SCGR 2003 Meeting, Montréal, QC, Canada, 6–9 July 2003. [Google Scholar]
- Helrick, K. (Ed.) AOAC method 942.05—Ash in Animal Feeds. In Official Method of Analysis of the Association of Official Analytical Chemists, 15th ed.; Association of Official Analytical Chemists: Arlington, VA, USA, 1990; p. 70. [Google Scholar]
- Helrick, K. (Ed.) AOAC method 973.18—Fiber (Acid Detergent) and Lignin in Animal Feeds. In Official Method of Analysis of the Association of Official Analytical Chemists, 15th ed.; Association of Official Analytical Chemists: Arlington, VA, USA, 1990; p. 82. [Google Scholar]
- Mani, S.; Tabil, L.G.; Sokhansanj, S. Effects of compressive force, particle size and moisture content on mechanical properties of biomass pellets from grasses. Biomass Bioenergy 2006, 30, 648–654. [Google Scholar] [CrossRef]
- Fagerlund, G. Determination of the Specific Surface Area of Solids by Gas Adsorption—BET Method, 2nd ed.; ISO: Geneva, Switzerland, 2010. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 61, 373–380. [Google Scholar] [CrossRef]
- Ilyas, R.A.; Sapuan, S.M.; Ishak, M.R.; Zainudin, E.S. Effect of Delignification on the Physical, Thermal, Chemical, and Structural Properties of Sugar Palm Fibre. Bioresources 2017, 12, 8734–8754. [Google Scholar]
- Segal, L.; Creely, J.J.; Martin, A.E.; Conard, C.M. An Empirical Method for Estimating the Degree of Crystallinity of Native Cellulose Using the X-Ray Diffractometer. Text. Res. J. 1959, 29, 786–794. [Google Scholar] [CrossRef]
- Karoyo, A.H.; Dehabadi, L.; Wilson, L.D. Renewable Starch Carriers with Switchable Adsorption Properties. ACS Sustain. Chem. Eng. 2018, 6, 4603–4613. [Google Scholar] [CrossRef]
- Bhatnagar, R.; Gupta, G.; Yadav, S. A Review on Composition and Properties of Banana Fibers. IJAER 2015, 6, 49–52. [Google Scholar]
- Wang, H.; Rostle, R.; Kessler, R.W. Removing Pectin and Lignin During Chemical Processing of Hemp for Textile Applications. Text. Res. J. 2003, 73, 664–669. [Google Scholar] [CrossRef]
- Roe, M.B.; Sniffen, C.J.; Chase, L.E. Techniques for Measuring Protein Fractions in Feedstuffs. In Proceedings—Cornell Nutrition Conference for Feed Manufacturers; Cornell University: Ithaca, NY, USA, 1990; pp. 81–88. [Google Scholar]
- Mahardika, M.; Abral, H.; Kasim, A.; Arief, S.; Asrofi, M. Production of Nanocellulose from Pineapple Leaf Fibers via High-Shear Homogenization and Ultrasonication. Fibers 2018, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Chan, F.; Chui, Y.-H.; Xiao, H. Characterization of flax fibers modified by alkaline, enzyme, and steam-heat treatments. BioResources 2012, 7, 4109–4121. [Google Scholar]
- Hosur, M.; Maroju, H.; Jeelani, S. Comparison of Effects of Alkali Treatment on Flax Fibre Reinforced Polyester and Polyester-biopolymer Blend Resins. Polym. Compos. 2015, 23, 229–242. [Google Scholar] [CrossRef]
- Dehabadi, L.; Wilson, L.D. Polysaccharide-Based Materials and Their Adsorption Properties in Aqueous Solution. Carbohydr. Polym. 2014, 113, 471–479. [Google Scholar] [CrossRef]
- Mohamed, M.H.; Wilson, L.D.; Headley, J.V. Design and characterization of novel β-cyclodextrin based copolymer materials. Carbohydr. Res. 2011, 346, 219–229. [Google Scholar] [CrossRef]
- Dong, Z.; Ding, R.-Y.; Zheng, L.; Zhang, X.; Yu, C.-W. Thermal Properties of Flax Fiber Scoured by Different Methods. Therm. Sci. 2015, 19, 939–945. [Google Scholar]
- Lee, S.G.; Choi, S.-S.; Park, W.H.; Cho, D. Characterization of surface modified flax fibers and their biocomposites with PHB. Macromol. Symp. 2003, 197, 89–99. [Google Scholar] [CrossRef]
- Saha, P.; Chowdhury, S.; Gupta, S.; Kumar, I.; Kumar, R. Assessment on the removal of malachite green using tamarind fruit shell as biosorbent. Clean Soil Air Water 2010, 38, 437–445. [Google Scholar] [CrossRef]
- Lazic, B.D.; Janjic, S.D.; Bijavec, T.; Kostic, M.M. Effect of chemical treatments on chemical composition and properties of flax fibers. J. Serb. Chem. Soc. 2016, 81, 1–16. [Google Scholar] [CrossRef]
- Haque, M.; Hasan, M.; Islam, S.; Ali, E. Physico-mechanical properties of chemically treated palm and coir fiber reinforced polypropylene composites. Bioresour. Technol. 2009, 100, 4903–4906. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.A.S.; Norton, A.; Newman, G. The water vapor sorption behavior of natural fibers. J. Appl. Polym. 2009, 112, 1524–1537. [Google Scholar] [CrossRef]
- Graaf, J.; Dillen, A.J.; Jong, K.P.; Koningsberger, D.C. Preparation of Highly Dispersed Pt Particles in Zeolite Y with a Narrow Particle Size Distribution: Characterization by Hydrogen Chemisorption, TEM, EXAFS Spectroscopy, and Particle Modeling. J. Catal. 2001, 203, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhu, H.; Njuguna, J.; Abhyankar, H. Recent Development of Flax Fibres and Their Reinforced Composites Based on Different Polymeric Matrices. Materials 2013, 6, 5171–5198. [Google Scholar] [CrossRef]
- Stevens, D.J.; Elton, G.A.H. Thermal Properties of the Starch Water System. Starch Starke 1971, 23, 8–11. [Google Scholar] [CrossRef]
- Aziz, S.H.; Ansell, M.P. The effect of alkalization and fibre alignment on the mechanical and thermal properties of kenaf and hemp bast fibre composites. Part 1. Polyester resin matrix. Compos. Sci. Technol. 2004, 64, 1219–1230. [Google Scholar] [CrossRef]
- Deepa, B.; Abraham, E.; Cherian, B.M.; Bismarck, A.; Blaker, J.J.; Pothan, L.A.; Leao, A.L.; de Souza, S.F.; Kottaisamy, M. Structure, morphology and thermal characteristics of banana nano fibers obtained by steam explosion. Bioresour. Technol. 2011, 102, 1988–1997. [Google Scholar] [CrossRef]
- Kabir, M.M.; Wang, H.; Cardona, F.; Aravinthan, T. Effect of chemical treatment on the mechanical and thermal properties of hemp fibre reinforced thermoset sandwich composites. In Incorporating Sustainable Practice in Mechanics and Structures of Materials; CRC Press: Boca Raton, FL, USA, 2010; pp. 439–444. [Google Scholar]
- Burikov, S.; Dolenko, T.; Patsaeva, S.; Starokurov, Y.; Yazhakov, V. Raman and IR Spectroscopy Research in Hydrogen Bonding of Water-Ethanol System. Mol. Phys. 2010, 108, 2427–2436. [Google Scholar] [CrossRef]
- Bellavia, G.; Paccou, L.; Achir, S.; Guinet, Y.; Siepmann, J.; Hedoux, A. Analysis of Bulk and Hydration Water During Thermal’ Lysozyme Denaturation Using Raman Scattering. Food Biophys. 2013, 8, 170–176. [Google Scholar] [CrossRef]
- Mundy, W.C.; Gutierrez, L.; Spedding, F.H. Raman intensities of the uncoupled OD oscillators in liquid water. J. Chem. Phys. 1973, 59, 2173–2182. [Google Scholar] [CrossRef]
- Panthapulakkal, S.; Sain, M. Isolation of Nano Fibres from Hemp and Flax and Their Thermoplastic Composites. Polym. Plast. Technol. Eng. 2013, 2, 9–16. [Google Scholar]
- Stamboulisa, A.; Baillie, C.A.; Peijs, T. Effects of environmental conditions on mechanical and physical properties of flax fibers. Compos. Part A Appl. Sci. Manuf. 2001, 32, 1105–1115. [Google Scholar] [CrossRef]
- Sorieul, M.; Dickson, A.; Hill, S.J.; Pearson, H. Plant Fibre: Molecular Structure and Biomechanical Properties, of a Complex Living Material, Influencing Its Deconstruction towards a Biobased Composite. Materials 2016, 9, 618. [Google Scholar] [CrossRef]
- Buchert, J.; Pere, J.; Johansson, L.-S.; Campbell, J.M. Analysis of the Surface Chemistry of Linen and Cotton Fabrics. Text. Res. J. 2001, 71, 626–629. [Google Scholar] [CrossRef]
- Tserki, V.; Zafeiropoulos, N.E.; Simon, F.; Panayiotou, C. A study of the effect of acetylation and propionylation surface treatments on natural fibres. Compos. Part A Appl. Sci. Manuf. 2005, 36, 1110–1118. [Google Scholar] [CrossRef]
- Wang, Q.; Xiao, S.; Shi, S.Q.; Cai, L. Mechanical property enhancement of self-bonded natural fiber material via controlling cell wall plasticity and structure. Mater. Des. 2019, 172, 1–8. [Google Scholar] [CrossRef]
- Kim, K.-M.; Oh, H.-T.; Lim, S.-J.; Ho, K.; Park, Y.; Lee, C.-H. Adsorption Equilibria of Water Vapor on Zeolite 3A, Zeolite 13X, and Dealuminated Y Zeolite. J. Chem. Eng. Data 2016, 61, 1547–1554. [Google Scholar] [CrossRef]
- Stana-Kleinschek, K.; Kreze, T.; Ribitsch, V.; Strnad, S. Reactivity and electrokinetical properties of different types of regenerated cellulose fibres. Colloid Surf. A Physicochem. Eng. Asp. 2001, 195, 275–284. [Google Scholar] [CrossRef]
- Mikhalovska, L.I.; Gunko, V.M.; Rugal, A.A.; Oranska, O.I.; Gornikov, Y.I.; Morvan, C.; Follain, N.; Domas, C.; Pakhlov, E.M.; Mikhalovsky, S.V. Cottonised flax fibres vs. cotton fibres: Structural, textural and adsorption characteristics. RSC Adv. 2012, 2, 2032–2042. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | LF (%) | CF (%) | PAF (%) |
|---|---|---|---|
| Hemicellulose | 14.5 | 11.4 | 18.9 |
| Cellulose | 70.3 | 85.1 | 75.7 |
| Lignin | 8.6 | 3.5 | 5.42 |
| Pectin | 2.0 | 0.0 | 0.0 |
| Sample Name | FWHM (N1s) | % (C1s) | FWHM (O1s) | % (O1s) | FWHM (N1s) | % (N1s) | O/C Ratio (%) |
|---|---|---|---|---|---|---|---|
| LF | 3.52 | 79.99 | 2.56 | 18.97 | 3.23 | 1.03 | 0.24 |
| CF | 3.96 | 74.23 | 2.99 | 24.61 | 2.75 | 1.16 | 0.33 |
| PAF | 3.95 | 76.47 | 4.00 | 21.87 | 3.44 | 1.66 | 0.26 |
| Parameters | LF | CF | PAF |
|---|---|---|---|
| Nitrogen adsorption (77 K) | |||
| Pore Size (nm) | 0.903 | 1.04 | 1.08 |
| Surface Area (m2/g) | 1.29 | 0.560 | 0.829 |
| Pore Volume (cm3/g) | 3.50 | 7.90 | 5.00 |
| KBET (L/g) | 5.34 | 7.15 | 4.80 |
| C | 1.15 | 1.11 | 1.22 |
| R2 | 0.993 | 0.996 | 0.994 |
| Reduced Chi-Sqr | 0.392 | 0.303 | 0.174 |
| Water vapour adsorption (298 K) | |||
| Qm (g/g) | 4.26 | 4.45 | 4.62 |
| (wt.%) | 25 | 29 | 21 |
| Surface Area (m2/g) | 153 | 159 | 167 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dehabadi, L.; Karoyo, A.H.; Soleimani, M.; Alabi, W.O.; Simonson, C.J.; Wilson, L.D. Flax Biomass Conversion via Controlled Oxidation: Facile Tuning of Physicochemical Properties. Bioengineering 2020, 7, 38. https://doi.org/10.3390/bioengineering7020038
Dehabadi L, Karoyo AH, Soleimani M, Alabi WO, Simonson CJ, Wilson LD. Flax Biomass Conversion via Controlled Oxidation: Facile Tuning of Physicochemical Properties. Bioengineering. 2020; 7(2):38. https://doi.org/10.3390/bioengineering7020038
Chicago/Turabian StyleDehabadi, Leila, Abdalla H. Karoyo, Majid Soleimani, Wahab O. Alabi, Carey J. Simonson, and Lee D. Wilson. 2020. "Flax Biomass Conversion via Controlled Oxidation: Facile Tuning of Physicochemical Properties" Bioengineering 7, no. 2: 38. https://doi.org/10.3390/bioengineering7020038