Cutting Edge Therapeutic Insights Derived from Molecular Biology of Pediatric High-Grade Glioma and Diffuse Intrinsic Pontine Glioma (DIPG)

 ,
,
Abstract
:1. Pathology of Pediatric High-Grade Gliomas and Diffuse Intrinsic Pontine Gliomas (DIPG)
2. Molecular Alterations of pHGG and DIPG
3. Histone H3-K27M and G34R/V-Specific Mechanisms in pHGG
4. Standard of Care Therapies in pHGG including DIPG
5. Histone H3-K27M and G34R/V-Specific Therapies
6. Therapeutic Delivery
7. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2015, 16 (Suppl. 10), x1–x36. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Veldhuijzen van Zanten, S.E.M.; Colditz, N.; Baugh, J.; Chaney, B.; Hoffmann, M.; Lane, A.; Fuller, C.; Miles, L.; Hawkins, C.; et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018, 36, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Merchant, T.E.; Pollack, I.F.; Loeffler, J.S. Brain tumors across the age spectrum: Biology, therapy, and late effects. Semin. Radiat. Oncol. 2010, 20, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Khatua, S.; Zaky, W. Diffuse intrinsic pontine glioma: Time for therapeutic optimism. CNS Oncol. 2014, 3, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J. Pediatric high grade glioma: A review and update on tumor clinical characteristics and biology. Front. Oncol. 2012, 2, 105. [Google Scholar] [CrossRef] [PubMed]
- Chamdine, O.; Gajjar, A. Molecular characteristics of pediatric high-grade gliomas. CNS Oncol. 2014, 3, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, D.A.; Mackay, A.; Little, S.E.; Carvalho, D.; Viana-Pereira, M.; Tamber, N.; Grigoriadis, A.E.; Ashworth, A.; Reis, R.M.; Ellison, D.W.; et al. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin. Cancer Res. 2010, 16, 3368–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, K.M.; Hoeman, C.M.; Becher, O.J. Children are not just little adults: Recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr. Res. 2014, 75, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32. [Google Scholar] [CrossRef] [PubMed]
- Vanan, M.I.; Eisenstat, D.D. Management of high-grade gliomas in the pediatric patient: Past, present, and future. Neurooncol. Pract. 2014, 1, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.; Severson, E.; Gay, L.; Vergilio, J.A.; Elvin, J.; Suh, J.; Daniel, S.; Covert, M.; Frampton, G.M.; Hsu, S.; et al. Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumor Mutational Burden, and Hypermutation Signatures. Oncologist 2017, 22, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Li, Y.; Xi, Y.; Jiang, S.; Stratton, S.; Peng, D.; Tanaka, K.; Ren, Y.; Xia, Z.; Wu, J.; et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 2014, 508, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakacki, R.I.; Cohen, K.J.; Buxton, A.; Krailo, M.D.; Burger, P.C.; Rosenblum, M.K.; Brat, D.J.; Hamilton, R.L.; Eckel, S.P.; Zhou, T.; et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: A report of the Children’s Oncology Group ACNS0423 study. Neuro Oncol. 2016, 18, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grill, J.; Massimino, M.; Bouffet, E.; Azizi, A.A.; McCowage, G.; Cañete, A.; Saran, F.; Le Deley, M.C.; Varlet, P.; Morgan, P.S.; et al. Phase II, Open-Label, Randomized, Multicenter Trial (HERBY) of Bevacizumab in Pediatric Patients With Newly Diagnosed High-Grade Glioma. J. Clin. Oncol. 2018, 36, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.W.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 2018, 33, 829–842.e5. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, X.; Geng, Y.; Bai, Q.; Pan, C.; Sun, Y.; Chen, X.; Yu, H.; Wu, Y.; Zhang, P.; et al. Patient-derived DIPG cells preserve stem-like characteristics and generate orthotopic tumors. Oncotarget 2017, 8, 76644–76655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Pan, C.-C.; Li, D. The historical change of brainstem glioma diagnosis and treatment: From imaging to molecular pathology and then molecular imaging. Chin. Neurosurg. J. 2015, 1, 4. [Google Scholar] [CrossRef]
- Hamisch, C.; Kickingereder, P.; Fischer, M.; Simon, T.; Ruge, M.I. Update on the diagnostic value and safety of stereotactic biopsy for pediatric brainstem tumors: A systematic review and meta-analysis of 735 cases. J. Neurosurg. Pediatr. 2017, 20, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Carai, A.; Mastronuzzi, A.; De Benedictis, A.; Messina, R.; Cacchione, A.; Miele, E.; Randi, F.; Esposito, G.; Trezza, A.; Colafati, G.S.; et al. Robot-Assisted Stereotactic Biopsy of Diffuse Intrinsic Pontine Glioma: A Single-Center Experience. World Neurosurg. 2017, 101, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Piunti, A.; Lulla, R.R.; Qi, J.; Horbinski, C.M.; Tomita, T.; James, C.D.; Shilatifard, A.; Saratsis, A.M. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol. Commun. 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Clymer, J.; Kieran, M.W. The Integration of Biology Into the Treatment of Diffuse Intrinsic Pontine Glioma: A Review of the North American Clinical Trial Perspective. Front. Oncol. 2018, 8, 169. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Marshall, L.; Perryman, L.; Bax, D.A.; Little, S.E.; Viana-Pereira, M.; Sharp, S.Y.; Vassal, G.; Pearson, A.D.; Reis, R.M.; et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010, 70, 9243–9252. [Google Scholar] [CrossRef] [PubMed]
- Kurscheid, S.; Bady, P.; Sciuscio, D.; Samarzija, I.; Shay, T.; Vassallo, I.; Criekinge, W.V.; Daniel, R.T.; van den Bent, M.J.; Marosi, C.; et al. Chromosome 7 gain and DNA hypermethylation at the HOXA10 locus are associated with expression of a stem cell related HOX-signature in glioblastoma. Genome Biol. 2015, 16, 16. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.M.; Smith, J.S.; Chen, Y.; Chen, J.; Phillips, H.S.; Aldape, K.D.; Zardo, G.; Nigro, J.; James, C.D.; Fridlyand, J.; et al. Reversing HOXA9 oncogene activation by PI3K inhibition: Epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res. 2010, 70, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Audia, J.E.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.E.; Brown, P.J.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, A.; Werner, T.; Chung, C.W.; Savitski, M.M.; Falth Savitski, M.; Grandi, P.; Hopf, C.; Lindon, M.; Neubauer, G.; Prinjha, R.K.; et al. The commonly used PI3-kinase probe LY294002 is an inhibitor of BET bromodomains. ACS Chem. Biol. 2014, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Maachani, U.B.; Schweitzer, M.; Singh, R.; Wang, M.; Chang, R.; Souweidane, M.M. Dual Inhibition of PI3K/AKT and MEK/ERK Pathways Induces Synergistic Antitumor Effects in Diffuse Intrinsic Pontine Glioma Cells. Transl. Oncol. 2017, 10, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, O.J.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; Haque, S.; De Braganca, K.C.; Kolesar, J.M.; Huse, J.T.; Modak, S.; Wexler, L.H.; et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016, 7, 11185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozono, D.; Li, J.; Nitta, M.; Sampetrean, O.; Gonda, D.; Kushwaha, D.S.; Merzon, D.; Ramakrishnan, V.; Zhu, S.; Zhu, K.; et al. Dynamic epigenetic regulation of glioblastoma tumorigenicity through LSD1 modulation of MYC expression. Proc. Natl. Acad. Sci. USA 2015, 112, E4055–E4064. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, W.; Zhu, J.; Wang, L.; Wu, X.; Shan, H. Combination of EZH2 inhibitor and BET inhibitor for treatment of diffuse intrinsic pontine glioma. Cell Biosci. 2017, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Cordero, F.J.; Huang, Z.; Grenier, C.; He, X.; Hu, G.; McLendon, R.E.; Murphy, S.K.; Hashizume, R.; Becher, O.J. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol. Cancer Res. 2017, 15, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Halvorson, K.G.; Barton, K.L.; Schroeder, K.; Misuraca, K.L.; Hoeman, C.; Chung, A.; Crabtree, D.M.; Cordero, F.J.; Singh, R.; Spasojevic, I.; et al. A high-throughput in vitro drug screen in a genetically engineered mouse model of diffuse intrinsic pontine glioma identifies BMS-754807 as a promising therapeutic agent. PLoS ONE 2015, 10, e0118926. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.S.; Sengupta, S.; Lee, K.; Hura, N.; Fuller, C.; DeWire, M.; Stevenson, C.B.; Fouladi, M.; Drissi, R. BMI-1 is a potential therapeutic target in diffuse intrinsic pontine glioma. Oncotarget 2017, 8, 62962–62975. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, H.M.; Morgan, M.; Gao, X.; Jackson, J.; Rickels, R.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Eissenberg, J.C.; Shilatifard, A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 2014, 345, 1065–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingg, D.; Debbache, J.; Peña-Hernández, R.; Antunes, A.T.; Schaefer, S.M.; Cheng, P.F.; Zimmerli, D.; Haeusel, J.; Calçada, R.R.; Tuncer, E.; et al. EZH2-Mediated Primary Cilium Deconstruction Drives Metastatic Melanoma Formation. Cancer Cell 2018, 34. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Hattori, N.; Iida, N.; Yamashita, S.; Mori, A.; Kimura, K.; Yoshino, T.; Ushijima, T. Targeting of super-enhancers and mutant BRAF can suppress growth of BRAF-mutant colon cancer cells via repression of MAPK signaling pathway. Cancer Lett. 2017, 402, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Zakharia, Y.; Monga, V.; Swami, U.; Bossler, A.D.; Freesmeier, M.; Frees, M.; Khan, M.; Frydenlund, N.; Srikantha, R.; Vanneste, M.; et al. Targeting epigenetics for treatment of BRAF mutated metastatic melanoma with decitabine in combination with vemurafenib: A phase lb study. Oncotarget 2017, 8, 89182–89193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, Z.Z.; Muller, M.M.; Jain, S.U.; Allis, C.D.; Lewis, P.W.; Muir, T.W. Strategy for “detoxification” of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J. Am. Chem. Soc. 2014, 136, 13498–13501. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M.; Manton, C.A.; Bhat, K.P.; Tsai, W.W.; Aldape, K.; Barton, M.C.; Chandra, J. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol. 2011, 13, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.M.; Johnson, B.; Venkatarayan, A.; Flores, E.R.; Zhang, J.; Su, X.; Barton, M.; Lang, F.; Chandra, J. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro Oncol. 2015, 17, 1463–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drappatz, J.; Lee, E.Q.; Hammond, S.; Grimm, S.A.; Norden, A.D.; Beroukhim, R.; Gerard, M.; Schiff, D.; Chi, A.S.; Batchelor, T.T.; et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J. Neurooncol. 2012, 107, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L.; et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro Oncol. 2015, 17, 862–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, T.; Onar-Thomas, A.; Huang, J.; Lulla, R.; Fangusaro, J.; Kramer, K.; Baxter, P.; Fouladi, M.; Dunkel, I.J.; Warren, K.E.; et al. Dipg-22. A phase 1 trial of the histone deacetylase inhibitor panobinostat in pediatric patients with recurrent or refractory diffuse intrinsic pontine glioma: A pediatric brain tumor consortium (pbtc) study. Neuro Oncol. 2018, 20. [Google Scholar] [CrossRef]
- Masoudi, A.; Elopre, M.; Amini, E.; Nagel, M.E.; Ater, J.L.; Gopalakrishnan, V.; Wolff, J.E. Influence of valproic acid on outcome of high-grade gliomas in children. Anticancer Res. 2008, 28, 2437–2442. [Google Scholar] [PubMed]
- Subashi, E.; Cordero, F.J.; Halvorson, K.G.; Qi, Y.; Nouls, J.C.; Becher, O.J.; Johnson, G.A. Tumor location, but not H3.3K27M, significantly influences the blood-brain-barrier permeability in a genetic mouse model of pediatric high-grade glioma. J. Neurooncol. 2016, 126, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.C.; Chu, P.C.; Wang, H.Y.; Huang, C.Y.; Chen, P.Y.; Tsai, H.C.; Lu, Y.J.; Lee, P.Y.; Tseng, I.C.; Feng, L.Y.; et al. Focused ultrasound-induced blood-brain barrier opening to enhance temozolomide delivery for glioblastoma treatment: A preclinical study. PLoS ONE 2013, 8, e58995. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.H.; Ting, C.Y.; Liu, H.L.; Huang, C.Y.; Hsieh, H.Y.; Yen, T.C.; Wei, K.C.; Yeh, C.K. Antiangiogenic-targeting drug-loaded microbubbles combined with focused ultrasound for glioma treatment. Biomaterials 2013, 34, 2142–2155. [Google Scholar] [CrossRef] [PubMed]
- Peiris, P.M.; Abramowski, A.; McGinnity, J.; Doolittle, E.; Toy, R.; Gopalakrishnan, R.; Shah, S.; Bauer, L.; Ghaghada, K.B.; Hoimes, C.; et al. Treatment of Invasive Brain Tumors Using a Chain-like Nanoparticle. Cancer Res. 2015, 75, 1356–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Absinta, M.; Ha, S.K.; Nair, G.; Sati, P.; Luciano, N.J.; Palisoc, M.; Louveau, A.; Zaghloul, K.A.; Pittaluga, S.; Kipnis, J.; et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.Z.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2018, 6, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.; Romero, M.; Han, R.; Larson, J.; Becher, O.; Lee, D.; Monje, M.; Gopalakrishnan, V.; Zaky, W.; Chandra, J. Immu-19. LSD1 modulates nk cell immunotherapy through an onco-immunogenic gene signature in dipg. Neuro Oncol. 2018, 20. [Google Scholar] [CrossRef]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Baron, O.; Förthmann, B.; Lee, Y.W.; Terranova, C.; Ratzka, A.; Stachowiak, E.K.; Grothe, C.; Claus, P.; Stachowiak, M.K. Cooperation of nuclear fibroblast growth factor receptor 1 and Nurr1 offers new interactive mechanism in postmitotic development of mesencephalic dopaminergic neurons. J. Biol. Chem. 2012, 287, 19827–19840. [Google Scholar] [CrossRef] [PubMed]
- Choubey, L.; Collette, J.C.; Smith, K.M. Quantitative assessment of fibroblast growth factor receptor 1 expression in neurons and glia. Peerj 2017, 5, e3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funa, K.; Sasahara, M. The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J. Neuroimmune Pharmacol. 2014, 9, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Toho, T.; Nonoguchi, N.; Furuse, M.; Kuwabara, H.; Yoritsune, E.; Kawabata, S.; Kuroiwa, T.; Miyatake, S. The roles of platelet-derived growth factors and their receptors in brain radiation necrosis. Radiat. Oncol. 2014, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, M.; Shore, E.M. Common mutations in ALK2/ACVR1, a multi-faceted receptor, have roles in distinct pediatric musculoskeletal and neural orphan disorders. Cytokine Growth Factor Rev. 2016, 27, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]


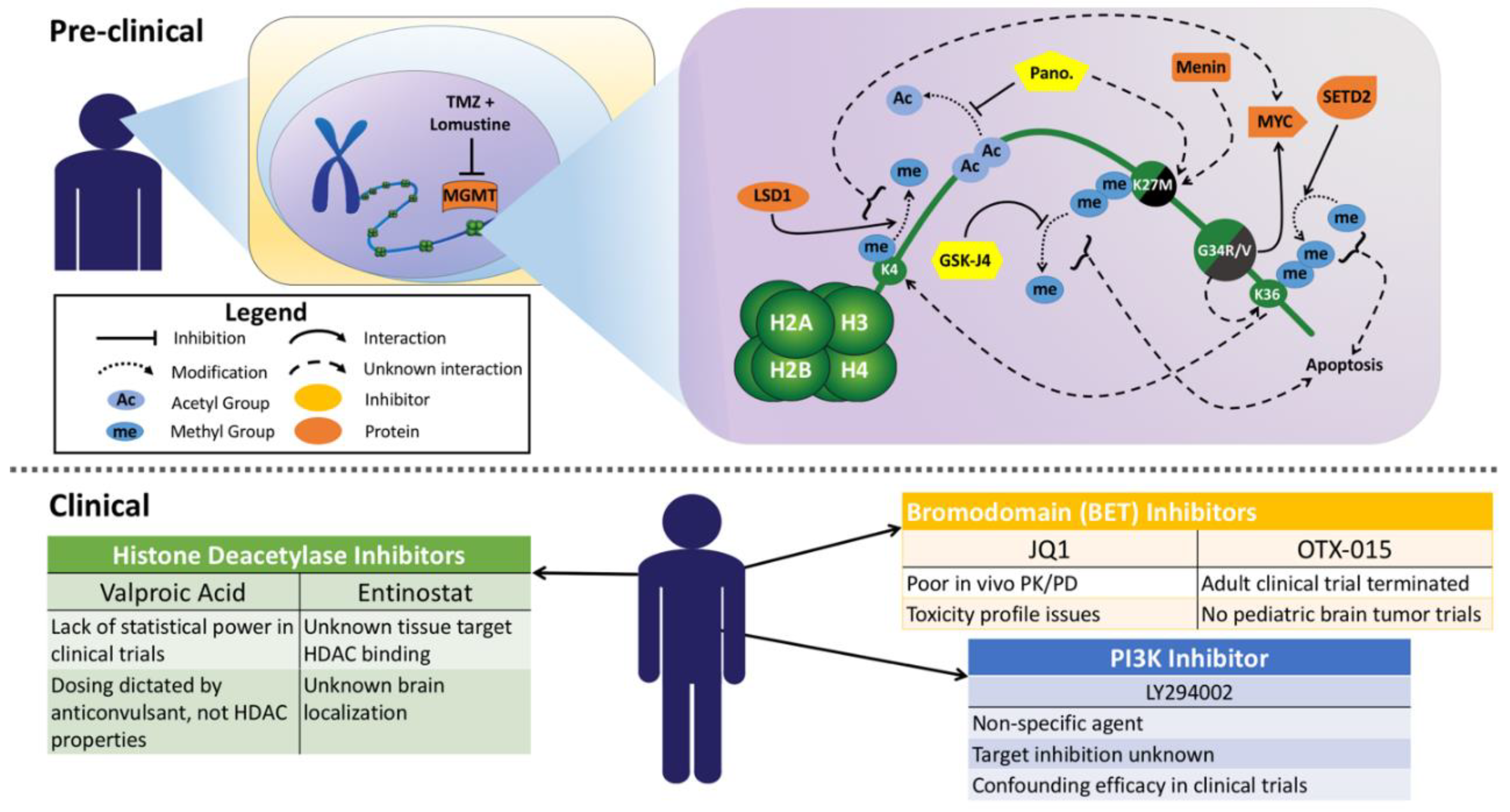{kind=link}
| Pediatric High-Grade Gliomas: Mutations, Features, and Novel Clinical Therapies | ||||
|---|---|---|---|---|
| Anatomical Classification | Defining Mutations | Features | Other Mutations | Targeted Therapies and Pediatric Clinical Trials |
Midline location |
|
|
|
|
|
|
| ||
|
|
| ||
Hemispheric location |
|
|
|
|
|
|
| ||
|
| |||
|
|
| ||
| Pre-Clinical Therapies for Pediatric High-Grade Gliomas | |||
|---|---|---|---|
| Compound(s) | Target | Rationale | Reference |
| VX-689 (renamed MK-5108) | AURKA (Aurora kinase A) | Destabilizes MYC | [48] |
| MI-2 | MEN1 (Menin) | Blocks menin-MLL-AF9 initiated leukemic oncogenesis; exact role in glioma undefined | [50,51] |
| GSK-J4 | JMJD3 (Jumonji-domain demethylase) | Prevents further demethylation of H3K27 mark in H3-K27M mutated glioma | [52] |
| OTX-015 JQ1 | BRD2/3/4 (Bromodomain-containing proteins) | Interrupts BRD → H3-acetylation binding that is increased by H3.3-K27M | [53,54,55] |
| GSK126 GSK343 EPZ-6438 | EZH2 (Enhancer of zeste homolog 2) | De-represses tumor suppressor p16INK4a and induces apoptosis | [55,56,57] |
| BMS-754807 | Multi-kinase, most potent against IGF-1R (Insulin-like growth factor 1 receptor) | Effective in compound screen; multiple valid targets in DIPG | [58] |
| PTC-209 | BMI-1 (polycomb group RING finger protein 4) | Induces cell cycle arrest and telomerase downregulation, reduces migration, increases sensitivity to radiotherapy | [59,60] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailey, C.P.; Figueroa, M.; Mohiuddin, S.; Zaky, W.; Chandra, J. Cutting Edge Therapeutic Insights Derived from Molecular Biology of Pediatric High-Grade Glioma and Diffuse Intrinsic Pontine Glioma (DIPG). Bioengineering 2018, 5, 88. https://doi.org/10.3390/bioengineering5040088
Bailey CP, Figueroa M, Mohiuddin S, Zaky W, Chandra J. Cutting Edge Therapeutic Insights Derived from Molecular Biology of Pediatric High-Grade Glioma and Diffuse Intrinsic Pontine Glioma (DIPG). Bioengineering. 2018; 5(4):88. https://doi.org/10.3390/bioengineering5040088
Chicago/Turabian StyleBailey, Cavan P., Mary Figueroa, Sana Mohiuddin, Wafik Zaky, and Joya Chandra. 2018. "Cutting Edge Therapeutic Insights Derived from Molecular Biology of Pediatric High-Grade Glioma and Diffuse Intrinsic Pontine Glioma (DIPG)" Bioengineering 5, no. 4: 88. https://doi.org/10.3390/bioengineering5040088