
A New Phenolic Acid Decarboxylase from the Brown-Rot Fungus Neolentinus lepideus Natively Decarboxylates Biosourced Sinapic Acid into Canolol, a Bioactive Phenolic Compound
 , , ,
, , ,
Abstract
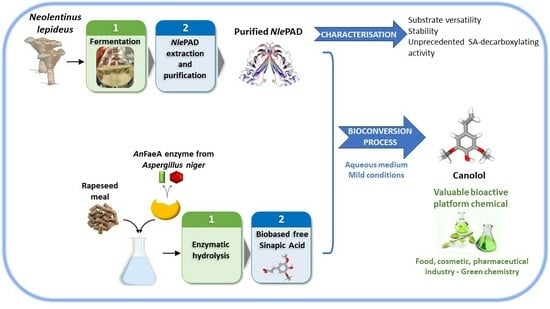
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Microorganisms and Culture Conditions
2.3. N. lepideus BRFM15 Intracellular Proteome Analysis
2.4. NlePAD Purification
2.5. Assay for PAD Activity
2.6. Assay for AnFaeA Activity
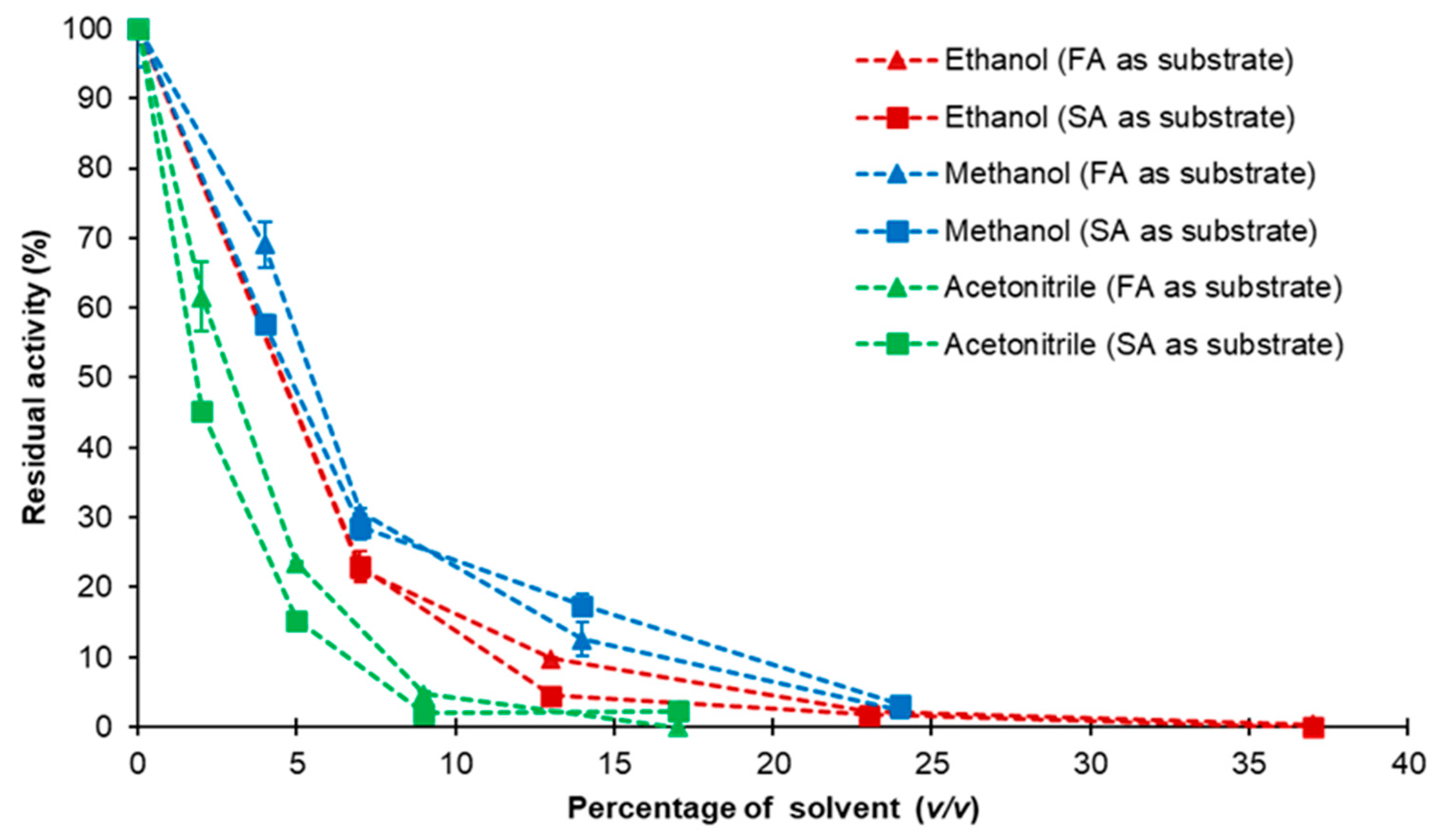
2.7. Effect of pH, Temperature and Organic Solvents on NlePAD Activity and Stability
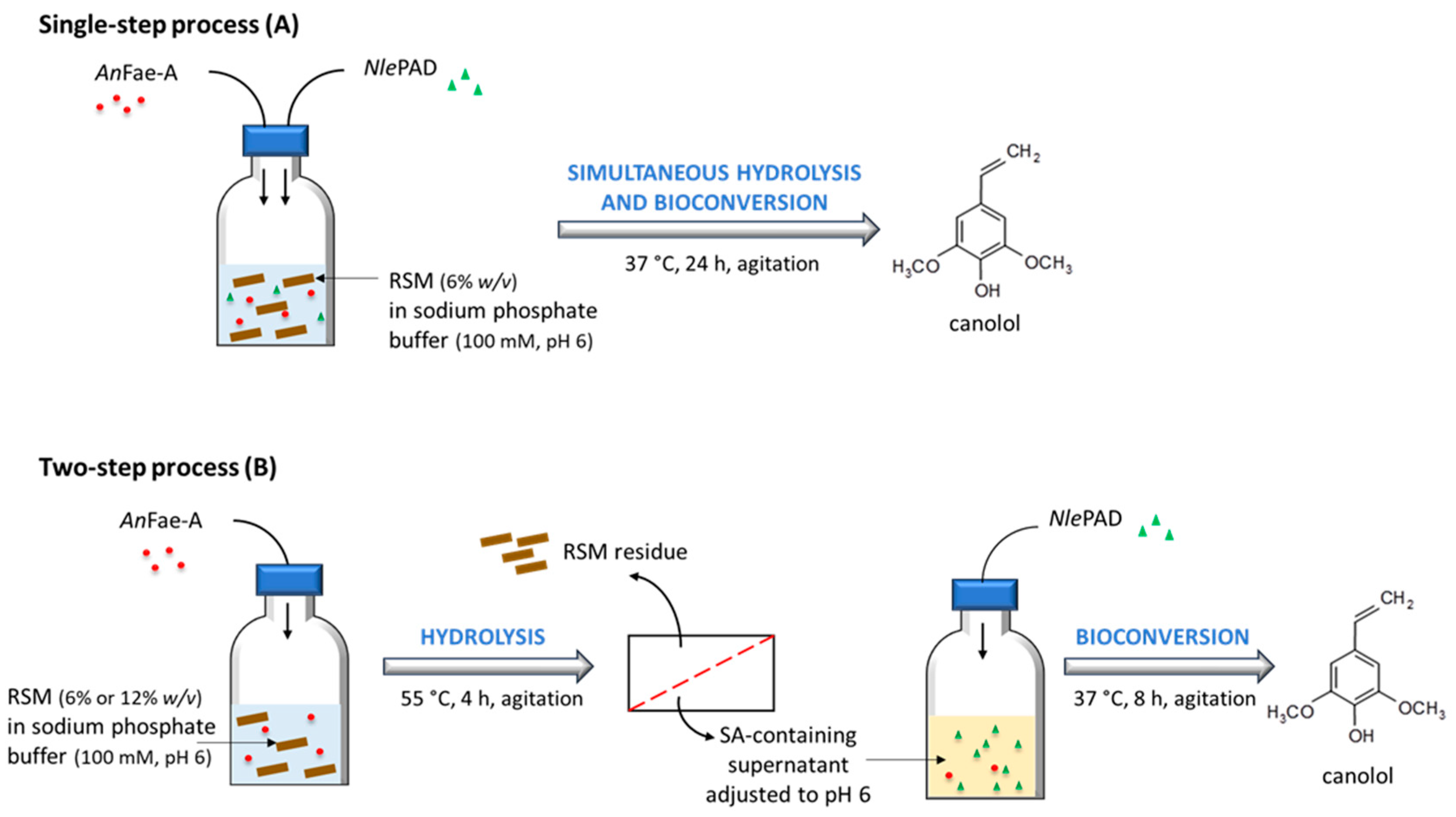
2.8. In Vitro Bioconversion of Commercial SA into Canolol
2.9. In Vitro Bioconversion of Biosourced SA from RSM into Canolol
2.10. High Performance Liquid Chromatography (HPLC) Analysis of the Monomeric Phenolics from NlePAD Reaction Medium
2.11. Bioinformatic Analysis
3. Results
3.1. Detection and Identification of the PAD from N. lepideus BRFM15 after Proteomic Analysis
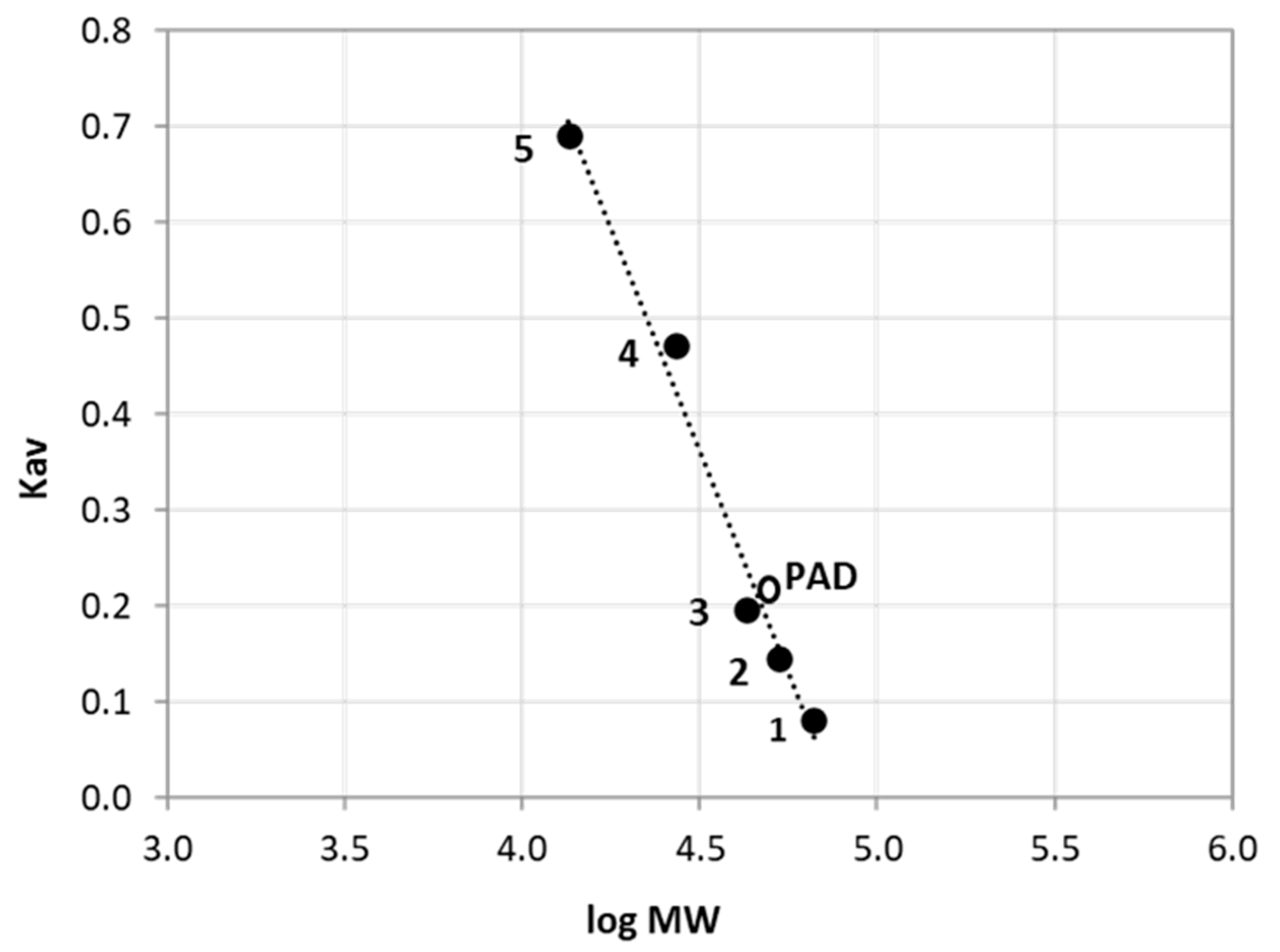
3.2. Purification of NlePAD
3.3. NlePAD Characterization
3.4. Comparison of the Predicted Structure of NlePAD with Bacterial PADs Shows Differences in Active Site
3.5. PAD-Catalyzed Bioconversion of Commercial and Biosourced SA into Canolol
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CafA | caffeic acid |
| pCA | p-coumaric acid |
| DDM | defatted dry matter |
| FA | ferulic acid |
| pHCA | p-hydroxycinnamic acid |
| LC-MS/MS | liquid chromatography–tandem mass spectrometry |
| PAD | phenolic acid decarboxylase |
| PES | polyethersulfone |
| PMSF | phenylmethanesulfonyl fluoride |
| RSM | rapeseed meal |
| SA | sinapic acid |
| SEC | size exclusion chromatography |
| TCA | trichloroacetic acid |
| 4-VG | 4-vinylguaiacol |
| 4-VP | 4-vinylphenol |
References
- Index Mundi. Agricultural Production, Supply, and Distribution. Available online: http://www.indexmundi.com/agriculture (accessed on 5 December 2023).
- Lomascolo, A.; Uzan-Boukhris, E.; Sigoillot, J.-C.; Fine, F. Rapeseed and sunflower meal: A review on biotechnology status and challenges. Appl. Microbiol. Biotechnol. 2012, 95, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Di Lena, G.; Sanchez Del Pulgar, J.; Lucarini, M.; Durazzo, A.; Ondrejíčková, P.; Oancea, F.; Frincu, R.M.; Aguzzi, A.; Ferrari Nicoli, S.; Casini, I.; et al. Valorization Potentials of Rapeseed Meal in a Biorefinery Perspective: Focus on Nutritional and Bioactive Components. Molecules 2021, 26, 6787. [Google Scholar] [CrossRef] [PubMed]
- Nehmeh, M.; Rodriguez-Donis, I.; Cavaco-Soares, A.; Evon, P.; Gerbaud, V.; Thiebaud-Roux, S. Bio-Refinery of Oilseeds: Oil Extraction, Secondary Metabolites Separation towards Protein Meal Valorisation—A Review. Processes 2022, 10, 841. [Google Scholar] [CrossRef]
- Wongsirichot, P.; Gonzalez-Miquel, M.; Winterburn, J. Recent advances in rapeseed meal as alternative feedstock for industrial biotechnology. Biochem. Eng. J. 2022, 180, 108373. [Google Scholar] [CrossRef]
- Baumert, A.; Milkowski, C.; Schmidt, J.; Nimtz, M.; Wray, V.; Strack, D. Formation of a complex pattern of sinapate esters in Brassica napus seeds, catalyzed by enzymes of a serine carboxypeptidase-like acyltransferase family? Phytochemistry 2005, 66, 1334–1345. [Google Scholar] [CrossRef]
- Milkowski, C.; Strack, D. Sinapate esters in brassicaceous plants: Biochemistry, molecular biology, evolution and metabolic engineering. Planta 2010, 232, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Siger, A.; Czubinski, J.; Dwiecki, K.; Kachlicki, P.; Nogala-Kalucka, M. Identification and antioxidant activity of sinapic acid derivatives in Brassica napus L. seed meal extracts: Main phenolic compounds in rapeseed. Eur. J. Lipid Sci. Technol. 2013, 115, 1130–1138. [Google Scholar] [CrossRef]
- Lomascolo, A.; Odinot, E.; Villeneuve, P.; Lecomte, J. Challenges and advances in biotechnological approaches for the synthesis of canolol and othervinylphenols from biobased p-hydroxycinnamic acids: A review. Biotechnol. Biofuels Bioprod. 2023, 16, 173. [Google Scholar] [CrossRef]
- Kamiyama, M.; Horiuchi, M.; Umano, K.; Kondo, K.; Otsuka, Y.; Shibamoto, T. Antioxidant/Anti-Inflammatory Activities and Chemical Composition of Extracts from the Mushroom Trametes versicolor. Int. J. Nutr. Food Sci. 2013, 2, 85–91. [Google Scholar] [CrossRef]
- Koski, A.; Pekkarinen, S.; Hopia, A.; Wähälä, K.; Heinonen, M. Processing of rapeseed oil: Effects on sinapic acid derivative content and oxidative stability. Eur. Food Res. Technol. 2003, 217, 110–114. [Google Scholar] [CrossRef]
- Wakamatsu, D.; Morimura, S.; Sawa, T.; Kida, K.; Nakai, C.; Maeda, H. Isolation, identification, and structure of a potent alkyl-peroxyl radical scavenger in crude canola oil, canolol. Biosci. Biotechnol. Biochem. 2005, 69, 1568–1574. [Google Scholar] [CrossRef]
- Galano, A.; Francisco-Marquez, M.; Alvarez-Idaboy, J.R. Canolol: A promising chemical agent against oxidative stress. J. Phys. Chem. 2011, 15, 8590–8596. [Google Scholar] [CrossRef]
- Maeda, H.; Tsukamoto, T.; Tatematsu, M. Anti-Inflammatory Agent and Cancer-Preventive Agent Comprising Canolol or Prodrug Thereof and Pharmaceutical, Cosmetic and Food Comprising the Same. U.S. Patent 20122/0122995 A1, 20 January 2012. [Google Scholar]
- Aouf, C.; Lecomte, J.; Villeneuve, P.; Dubreucq, E.; Fulcrand, H. Chemo-enzymatic functionalization of gallic and vanillic acids: Synthesis of bio-based epoxy resins prepolymers. Green Chem. 2012, 14, 2328–2336. [Google Scholar] [CrossRef]
- Zago, E.; Dubreucq, E.; Lecomte, J.; Villeneuve, P.; Fine, F.; Fulcrand, H.; Aouf, C. Synthesis of bio-based epoxy monomers from natural allyl- and vinyl phenols and the estimation of their affinity to the estrogen receptor α by molecular docking. New J. Chem. 2016, 40, 7701–7710. [Google Scholar] [CrossRef]
- Steinke, R.D.; Paulson, M.C. The production of steam-volatile phenols during the cooking and alcoholic fermentation of grain. J. Agric. Food Chem. 1964, 12, 381–387. [Google Scholar] [CrossRef]
- Huang, Z.; Dostal, L.; Rosazza, J.P. Purification and characterization of a ferulic acid decarboxylase from Pseudomonas fluorescens. J. Bacteriol. 1994, 176, 5912–5918. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-Y.; Volm, T.G.; Rosazza, J.P.N. Decarboxylation o ferulic acid to 4-vinylguaiacol by Bacillus pumilus in aqueous-organic solvent two-phase systems. Enzyme Microb. Technol. 1998, 23, 261–266. [Google Scholar] [CrossRef]
- Godoy, L.; Martinez, C.; Carrasco, N.; Ganga, M.A. Purification and characterization of p-coumarate decarboxylase and a vinylphenol reductase from Brettnomyces bruxellensis. Int. J. Food Microbiol. 2008, 127, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Li, X.; Huang, J.; Duan, Y.; Meng, Z.; Zhang, K.Q.; Yang, J. Cloning, sequencing, and overexpression in Escherichia coli of Enterobacter sp. Px6-4 gene for ferulic acid decarboxylase. Appl. Microbiol. Biotechnol. 2011, 89, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Landete, J.M.; Rodríguez, H.; Curiel, J.A.; de las Rivas, B.; Mancheño, J.M.; Muñoz, R. Gene cloning, expression, and characterization of phenolic acid decarboxylase from Lactobacillus brevis RM84. J. Ind. Microbiol. Biotechnol. 2012, 37, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Bhuiya, M.W.; Lee, S.G.; Jez, J.M.; Yu, O. Structure and mechanism of ferulic acid decarboxylase (FDC1) from Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2015, 81, 4216–4223. [Google Scholar] [CrossRef]
- Hu, H.; Li, L.; Ding, S. An organic solvent-tolerant phenolic acid decarboxylase from Bacillus licheniformis for the efficient bioconversion of hydroxycinnamic acids to vinyl phenol derivatives. Appl. Microbiol. Biotechnol. 2015, 99, 5071–5081. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; Grosse, F.; Bergeron, H.; Abokitse, K.; Lau, P.C.K. Structural analysis of Bacillus pumilus acid decarboxylase, a lipocalin-fold enzyme. Acta Cryst. Sect. F Struct. Biol. Cryst. Comm. 2010, F66, 1407–1414. [Google Scholar] [CrossRef]
- Rodriguez, H.; Angulo, I.; de las Rivas, B.; Campillo, N.; Páez, J.A.; Muňoz, R.; Mancheňo, J.M. p-Coumaric acid decarboxylase from Lactobacillus plantarum: Structural insights into the active site and decarboxylation catalytic mechanism. Proteins 2010, 78, 1662–1676. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.; Eborall, W.; Hyde, R.; Hart, S.; Turkenburg, J.P.; Grogan, G. Mutational analysis of phenolic acid decarboxylase from Bacillus subtilis (BsPAD), which converts bio-derived phenolic acids to styrene derivatives. Catal. Sci. Technol. 2012, 2, 1568–1574. [Google Scholar] [CrossRef]
- Pudel, F.; Habicht, V.; Piofczyk, T.; Matthäus, B.; Quirin, K.W.; Cawelius, A. Fluidized bed treatment of rapeseed meal and cake as possibility for the production of canolol. Oilseeds Fats Crops Lipids 2014, 21, 103. [Google Scholar] [CrossRef]
- Yang, M.; Zheng, C.; Zhou, Q.; Liu, C.; Li, W.; Huang, F. Influence of microwaves treatment of rapeseed on phenolic compounds and canolol content. J. Agric. Food Chem. 2014, 62, 1956–1963. [Google Scholar] [CrossRef] [PubMed]
- Zago, E.; Lecomte, J.; Barouh, N.; Aouf, C.; Carré, P.; Fine, F.; Villeneuve, P. Influence of rapeseed meal treatments on its total phenolic content and composition in sinapine, sinapic acid and canolol. Ind. Crops Prod. 2015, 76, 1061–1070. [Google Scholar] [CrossRef]
- Li, J.; Guo, Z. Concurrent extraction and transformation of bioactive phenolic compounds from rapeseed meal using pressurized solvent extraction system. Ind. Crop Prod. 2016, 94, 152–159. [Google Scholar] [CrossRef]
- Cavin, J.F.; Dartois, V.; Divies, C. Gene cloning, transcriptional analysis, purification, and characterization of phenolic acid decarboxylase from Bacillus subtilis. Appl. Environ. Microbiol. 1998, 64, 1466–1471. [Google Scholar] [CrossRef]
- Morley, K.L.; Grosse, S.; Leisch, H.; Lau, P.C.K. Antioxidant canolol production from a renewable feedstock via an engineered decarboxylase. Green Chem. 2013, 15, 3312–3317. [Google Scholar] [CrossRef]
- Li, Q.; Xia, Y.; Zhao, T.; Gong, Y.; Fang, S.; Chen, M. Improving the catalytic characteristics of phenolic acid decarboxylase from Bacillus amyloquefaciens by the engineering of N-terminus and C-terminus. BMC Biotechnol. 2021, 21, 44. [Google Scholar] [CrossRef]
- Xie, X.G.; Huang, C.Y.; Fu, W.Q.; Dai, C.C. Potential of endophytic fungus Phomopsis liquidambari for transformation and degradation of recalcitrant pollutant sinapic acid. Fungal Biol. 2016, 120, 402–413. [Google Scholar] [CrossRef]
- Linke, D.; Riemer, S.J.L.; Schimanski, S.; Nieter, A.; Krings, U.; Berger, R.G. Cold generation of smoke flavour by the first phenolic acid decarboxylase from a filamentous ascomycete—Isaria farinosa. Fungal Biol. 2017, 121, 763–774. [Google Scholar] [CrossRef]
- Maeda, M.; Tokashiki, M.; Tokashiki, M.; Uechi, K.; Ito, S.; Taira, T. Characterization and induction of phenolic acid decarboxylase from Aspergillus luchuensis. J. Sci. Bioeng. 2018, 126, 162–168. [Google Scholar] [CrossRef]
- Detering, T.; Mundry, K.; Berger, R.G. Generation of 4-vinylguaiacol through a novel high affinity ferulic acid decarboxylase to obtain smoke flavours without carcinogenic contaminants. PLoS ONE 2020, 15, e0244290. [Google Scholar] [CrossRef]
- JGI Mycocosm. The Fungal Genomics Resource. Available online: https://genome.jgi.doe.gov/programs/fungi/index.jsf (accessed on 5 December 2021).
- Lomascolo, A.; Odinot, E.; Sigoillot, J.-C.; Navarro, D.; Peyronnet, C.; Fine, F. Process for Preparing a Vinylphenolic Compound from a Precursor Hydroxycinnamic Acid Derived from an Oilseed Cake. International Patent WO2017/072450A1, 4 May 2017. [Google Scholar]
- Odinot, E.; Fine, F.; Sigoillot, J.C.; Navarro, D.; Laguna, O.; Bisotto, A.; Peyronnet, C.; Ginies, C.; Lecomte, J.; Faulds, C.B.; et al. A two-step bioconversion process for canolol production from rapeseed meal combining an Aspergillus niger feruloyl esterase and the fungus Neolentinus lepideus. Microorganisms. 2017, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Record, E.; Asther Mi Sigoillot, C.; Pages, S.; Punt, P.J.; Delattre, M.; Haon, M.; Van der Hondel, C.A.M.J.J.; Sigoillot, J.-C.; Lesage-Meessen, L.; Asther, M. Overproduction of Aspergillus niger feruloyl esterase for pulp bleaching application. Appl. Microbiol. Biotechnol. 2003, 62, 349–355. [Google Scholar] [CrossRef]
- Couturier, M.; Navarro, D.; Olivé, C.; Chevret, D.; Haon, M.; Favel, A.; Lesage-Meessen, L.; Henrissat, B.; Coutinho, P.M.; Berrin, J.G. Post-genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis. BMC Genom. 2012, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Arfi, Y.; Chevret, D.; Henrissat, B.; Berrin, J.G.; Levasseur, A.; Record, E. Characterization of salt-adapted secreted lignocellulolytic enzymes from the mangrove fungus Pestalotiopsis sp. Nat. Commun. 2013, 4, 1810. [Google Scholar] [CrossRef] [PubMed]
- JGI Mycocosm. The Fungal Genomics Resource. Neolentinus lepideus. Available online: https://mycocosm.jgi.doe.gov/Neole1/Neole1.home.html (accessed on 5 December 2022).
- Nagy, L.G.; Riley, R.; Tritt, A.; Adam, C.; Daum, C.; Floudas, D.; Sun, H.; Yadav, J.S.; Pangilinan, J.; Larsson, K.H.; et al. Comparative Genomics of Early-Diverging Mushroom-Forming Fungi Provides Insights into the Origins of Lignocellulose Decay Capabilities. Mol. Biol. Evol. 2016, 33, 959–970. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Ralet, M.-C.; Faulds, C.B.; Williamson, G.; Thibaut, J.-F. Degradation of feruloylated oligosaccharides from sugar-beet pulp and wheat bran by ferulic acid esterases from Aspergillus niger. Carbohydr. Res. 1994, 263, 257–269. [Google Scholar] [CrossRef]
- Laguna, O.; Odinot, E.; Bisotto, A.; Baréa, B.; Villeneuve, P.; Sigoillot, J.-C.; Record, E.; Faulds, C.B.; Fine, F.; Lesage-Meessen, L.; et al. Release of phenolic acids from sunflower and rapeseed meals using different carboxylic esters hydrolases from Aspergillus niger. Ind. Crops Prod. 2019, 139, 111579. [Google Scholar] [CrossRef]
- Multiple Sequence Alignment by CLUSTALW. Available online: https://www.genome.jp/tools-bin/clustalw (accessed on 5 November 2023).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Hebditech, M.; Warwicker, J. Charge and hydrophobicity are key features in sequence-trained machine learning models for predicting the biophysical properties of clinical-stage antibodies. PeerJ 2019, 7, e8199. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol (accessed on 30 August 2023).
- Navarro, D.; Chaduli, D.; Taussac, S.; Lesage-Meessen, L.; Grisel, S.; Haon, M.; Callac, P.; Courtecuisse, R.; Decock, C.; Dupont, J.; et al. Large-scale phenotyping of 1,000 fungal strains for the degradation of non-natural, industrial compounds. Commun. Biol. 2021, 4, 871. [Google Scholar] [CrossRef]
- Shimazano, H. Investigations on lignins and lignification. Identification of a phenolic ester in the culture medium of Lentinus lepideus and the O-methylation of Methyl p-coumarate to Methyl p-methoxycinnamate in vivo. Arch. Biochem. Biophys. 1959, 83, 206–215. [Google Scholar] [CrossRef]
- Duncan, C.; Deverall, F. Degradation of Wood Preservatives by Fungi. Appl. Microbiol. 1964, 12, 57–62. [Google Scholar] [CrossRef]
- Okamoto, K.; Kanawaku, R.; Masumoto, M.; Yanase, H. Efficient xylose fermentation by the brown rot fungus Neolentinus lepideus. Enzyme Microb. Technol. 2012, 50, 96–100. [Google Scholar] [CrossRef]
- Okamoto, K.; Nakawaka, S.; Kanawaku, R.; Kawamura, S. Ethanol production from cheese whey and expired milk by the brown rot fungus Neolentinus lepideus. Fermentation 2019, 5, 49. [Google Scholar] [CrossRef]
- Lomascolo, A.; Stentelaire, C.; Lesage-Meessen, L.; Asther, M. Basidiomycetes as new biotechnological agents to generate natural aromatic flavours for the food industry. Trends Biotechnol. 1999, 17, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Alvarado, I.; Navarro, D.; Record, E.; Asther Mi Asther, M.; Lesage-Meessen, L. Fungal biotransformation of p-coumaric acid into caffeic acid by Pycnoporus cinnabarinus: An alternative for producing a strong natural antioxidant. World J. Microbiol. Biotechnol. 2003, 19, 157–160. [Google Scholar] [CrossRef]
- Barthelmebs, L.; Diviès, C.; Cavin, J.F. Expression in Escherichia coli of native and chimeric phenolic acid decarboxylases with modified enzymatic activities and method for screening recombinant E. coli strains expressing these enzymes. Appl. Environ. Microbiol. 2001, 67, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Degrassi, G.; Polvrino de Laureto, P.; Bruschi, C.V. Purification and characterization of ferulate and p-coumarate decarboxylase from Bacillus pumilus. Appl. Environ. Microbiol. 1995, 61, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Edlin, D.A.N.; Narbad, A.; Gasson, M.J.; Dickinson, J.R.; Lloyd, D. Purification and characterization of hydroxycinnamate decarboxylase from Brettanomyces anomalus. Enzyme Microb. Technol. 1998, 22, 232–239. [Google Scholar] [CrossRef]
- Huang, H.K.; Tokashiki, M.; Maeno, S.; Onaga, S.; Taira, T.; Ito, S. Purification and properties of phenolic acid decarboxylase from Candida guilliermondii. J. Ind. Microbiol. Biotechnol. 2012, 39, 55–62. [Google Scholar] [CrossRef]
- Li, L.; Long, L.; Ding, S. Bioproduction of high-concentration 4-vinylguaiacol using whole-cell catalysis harboring an organic solvent-tolerant phenolic acid decarboxylase from Bacillus atrophaeus. Front. Microbiol. 2019, 10, 1798. [Google Scholar] [CrossRef]
- Prim, N.; ·Pastor, F.I.J.; Diaz, P. Biochemical studies on cloned Bacillus sp. BP-7 phenolic acid decarboxylase PadA. Appl. Microbiol. Biotechnol. 2003, 63, 51–56. [Google Scholar] [CrossRef]
- Jung, D.H.; Choi, W.; Choi, K.Y.; Jung, E.; Yun, H.; Kazlauskas, R.J.; Kim, B.G. Bioconversion of p-coumaric acid to p-hydroxystyrene using phenolic acid decarboxylase from B. amyloliquefaciens in biphasic reaction system. Appl. Microbiol. Biotechnol. 2013, 97, 1501–1511. [Google Scholar] [CrossRef]
- Pesci, L.; Baydadr, M.; Glueck, S.; Faber, K.; Liese, A.; Kara, S. Development and scaling-up of the fragrance compound 4-ethylguaiacol synthesis via a two-step chemo-enzymatic reaction. Org. Process Res. Dev. 2017, 21, 85–93. [Google Scholar] [CrossRef]
- Punt, P.J.; Levasseur, A.; Visser, H.; Wery, J.; Record, E. Fungal protein production: Design and production of chimeric proteins. Annu. Rev. Microbiol. 2011, 65, 57–69. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purification Step | Volume (mL) | Protein Concentration (mg.mL−1) | Activity b (U.mL−1) | Total Activity b (U) | Specific Activity b (U.mg Proteins−1) | Yield (%) | Purification (-Fold) |
|---|---|---|---|---|---|---|---|
| Crude extract | 140 | 2.075 | 57.1 | 7995 | 27.52 | ||
| DEAE Sepharose Fast Flow a | 14 | 5.472 | 440.9 | 6172 | 80.57 | 77 | 2.9 |
| Sephacryl S-100HR a | 1.55 | 3.279 | 1819.0 | 2819 | 554.70 | 35 | 20.1 |
| Superdex 75 Prep Grade a | 4 | 0.204 | 441.8 | 1767 | 2161.78 | 22 | 78.6 |
| Substrate | ||
|---|---|---|
| Sinapic Acid | Ferulic Acid | |
| Temperature range of activity | 30–50 °C | 30–55 °C |
| Optimal temperature | 37 °C | 45 °C |
| Temperature stability | ||
| Half-life (h) at 4 °C | >120 | >120 |
| 30 °C | 91.6 | 90 |
| 37 °C | 64.6 | 58 |
| 45 °C | 28.2 | 18.3 |
| 55 °C | 12.4 | 10.2 |
| pH range of activity | 5.5–7.5 | 5.0–7.5 |
| Optimal pH | 6 | 6.0–6.5 |
| pH stability | ||
| Residual activity after 2 days (%) | ||
| pH 4 | 16 | 12 |
| pH 5 | 72 | 81 |
| pH 6 | 88 | 77 |
| pH 7 | 97 | 97 |
| pH 7.5 | 90 | 91 |
| pH 8 | 95 | 100 |
| Residual activity after 7 days (%) | ||
| pH 4 | 1 | 1 |
| pH 5 | 31 | 37 |
| pH 6 | 70 | 63 |
| pH 7 | 86 | 90 |
| pH 7.5 | 84 | 84 |
| pH 8 | 80 | 92 |
| KM (mM) | 3.9 | 2.6 |
| Vmax (U.mg−1) | 600 | 3735 |
| kcat (s−1) | 6.3 | 39.2 |
| kcat/KM (s−1.mM−1) | 1.6 | 14.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odinot, E.; Bisotto-Mignot, A.; Frezouls, T.; Bissaro, B.; Navarro, D.; Record, E.; Cadoret, F.; Doan, A.; Chevret, D.; Fine, F.; et al. A New Phenolic Acid Decarboxylase from the Brown-Rot Fungus Neolentinus lepideus Natively Decarboxylates Biosourced Sinapic Acid into Canolol, a Bioactive Phenolic Compound. Bioengineering 2024, 11, 181. https://doi.org/10.3390/bioengineering11020181
Odinot E, Bisotto-Mignot A, Frezouls T, Bissaro B, Navarro D, Record E, Cadoret F, Doan A, Chevret D, Fine F, et al. A New Phenolic Acid Decarboxylase from the Brown-Rot Fungus Neolentinus lepideus Natively Decarboxylates Biosourced Sinapic Acid into Canolol, a Bioactive Phenolic Compound. Bioengineering. 2024; 11(2):181. https://doi.org/10.3390/bioengineering11020181
Chicago/Turabian StyleOdinot, Elise, Alexandra Bisotto-Mignot, Toinou Frezouls, Bastien Bissaro, David Navarro, Eric Record, Frédéric Cadoret, Annick Doan, Didier Chevret, Frédéric Fine, and et al. 2024. "A New Phenolic Acid Decarboxylase from the Brown-Rot Fungus Neolentinus lepideus Natively Decarboxylates Biosourced Sinapic Acid into Canolol, a Bioactive Phenolic Compound" Bioengineering 11, no. 2: 181. https://doi.org/10.3390/bioengineering11020181