
Organotypic 3D Co-Culture of Human Pleura as a Novel In Vitro Model of Staphylococcus aureus Infection and Biofilm Development
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
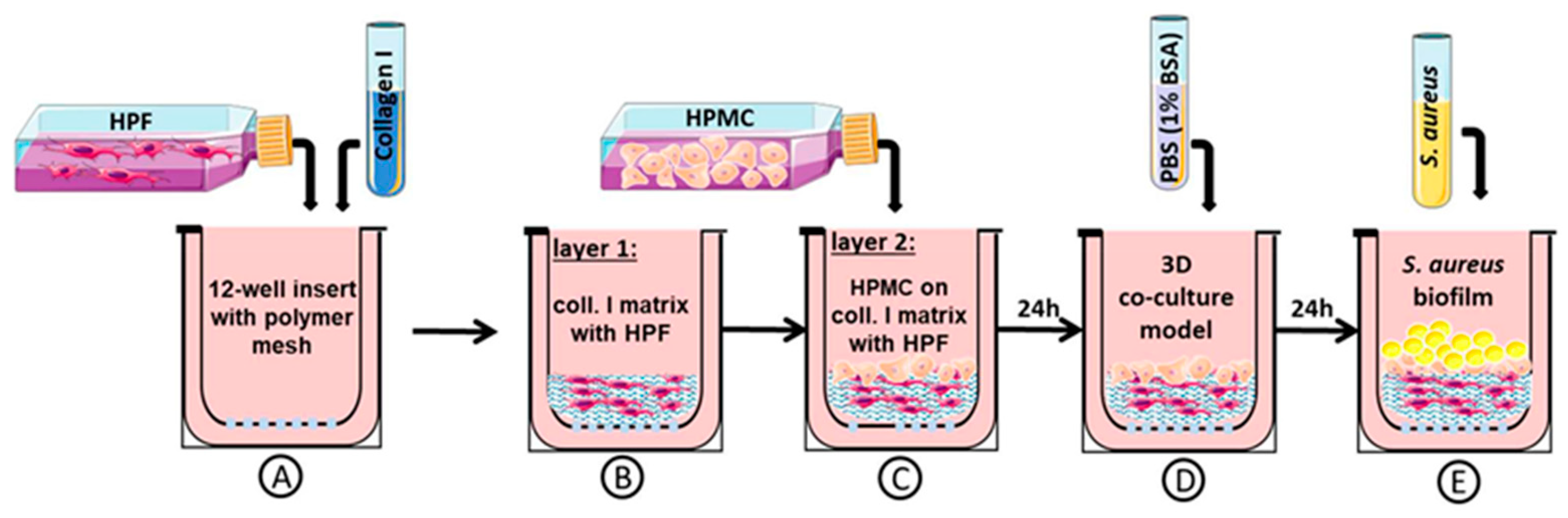
2.1. Three-Dimensional Organotypic Co-Culture Model of Pleura
2.2. Bacterial Strain
2.3. Three-Dimensional Organotypic Co-Culture Model of Pleura and Bacterial Infection
2.4. Histological Analysis and Immunostaining
2.5. Cytokine and Growth Factor Quantification
2.6. Barrier Integrity Assessment
2.7. Determination of Cell Viability
2.8. Characterization of Biofilm in the 3D Co-Culture Model of Pleura
2.9. Quantification of Penetration and Bacterial Accumulation
2.10. Statistical Analysis
3. Results
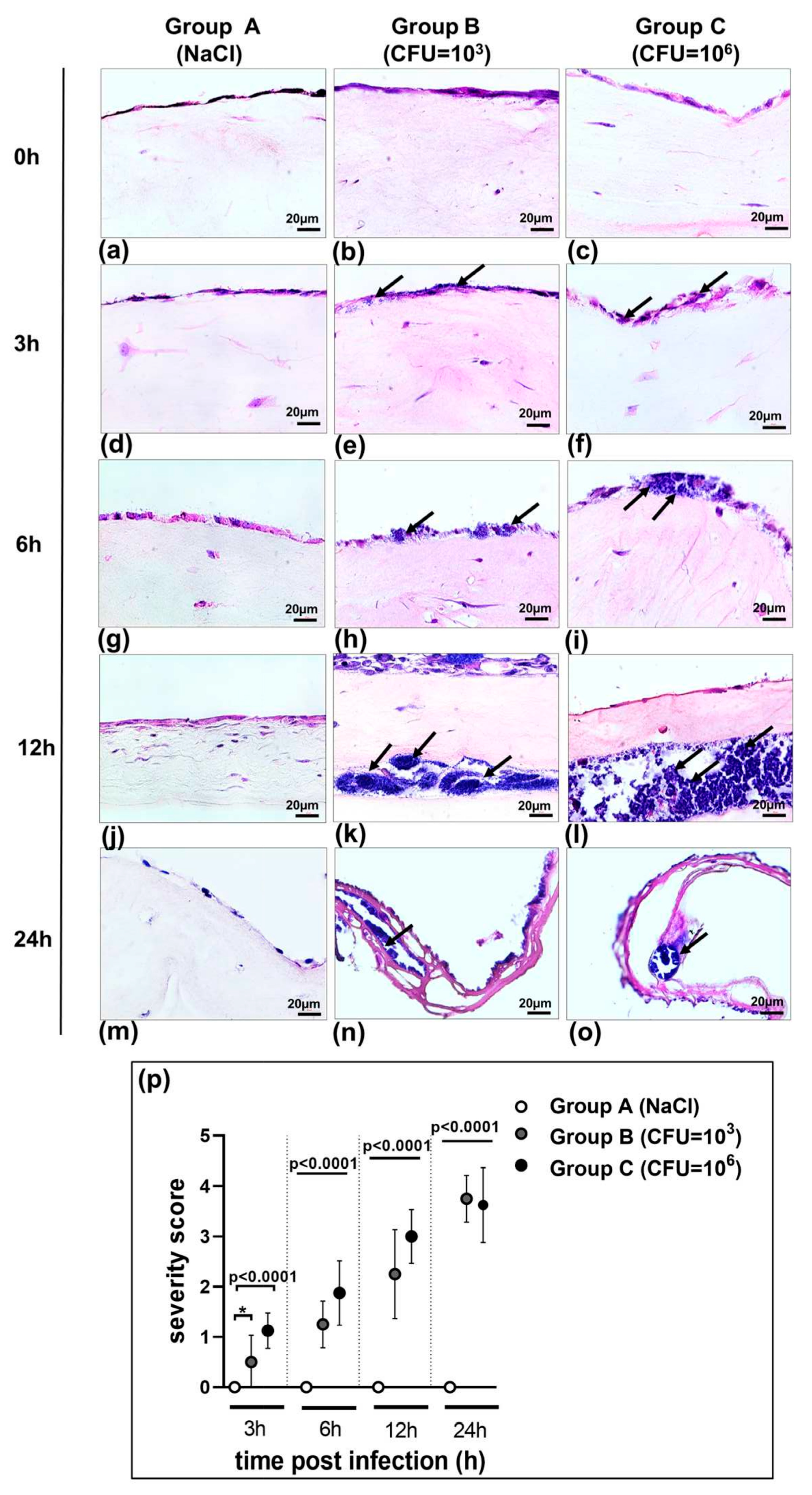
3.1. Morphology of 3D Organotypic Co-Culture Model of Pleura after Infection
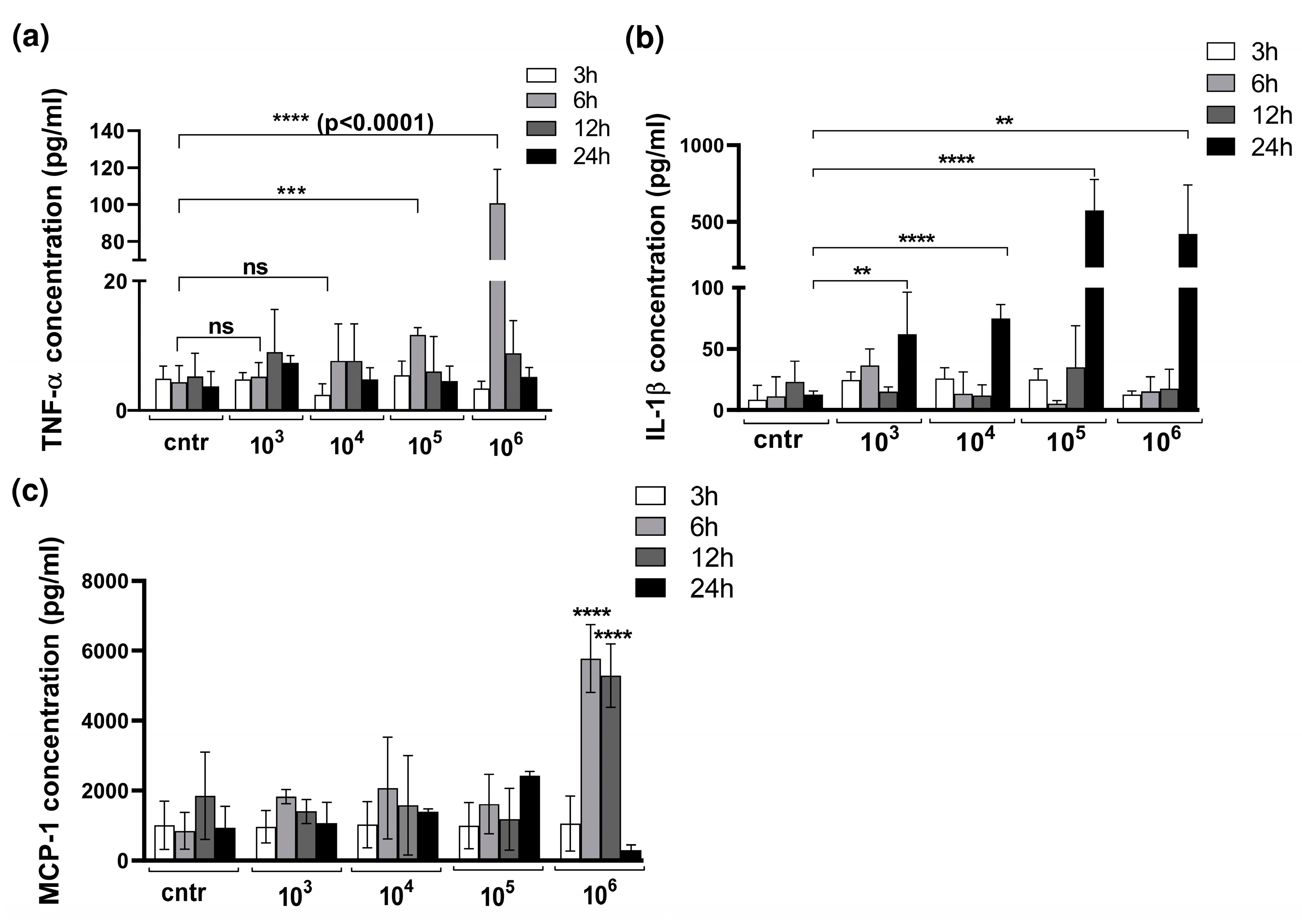
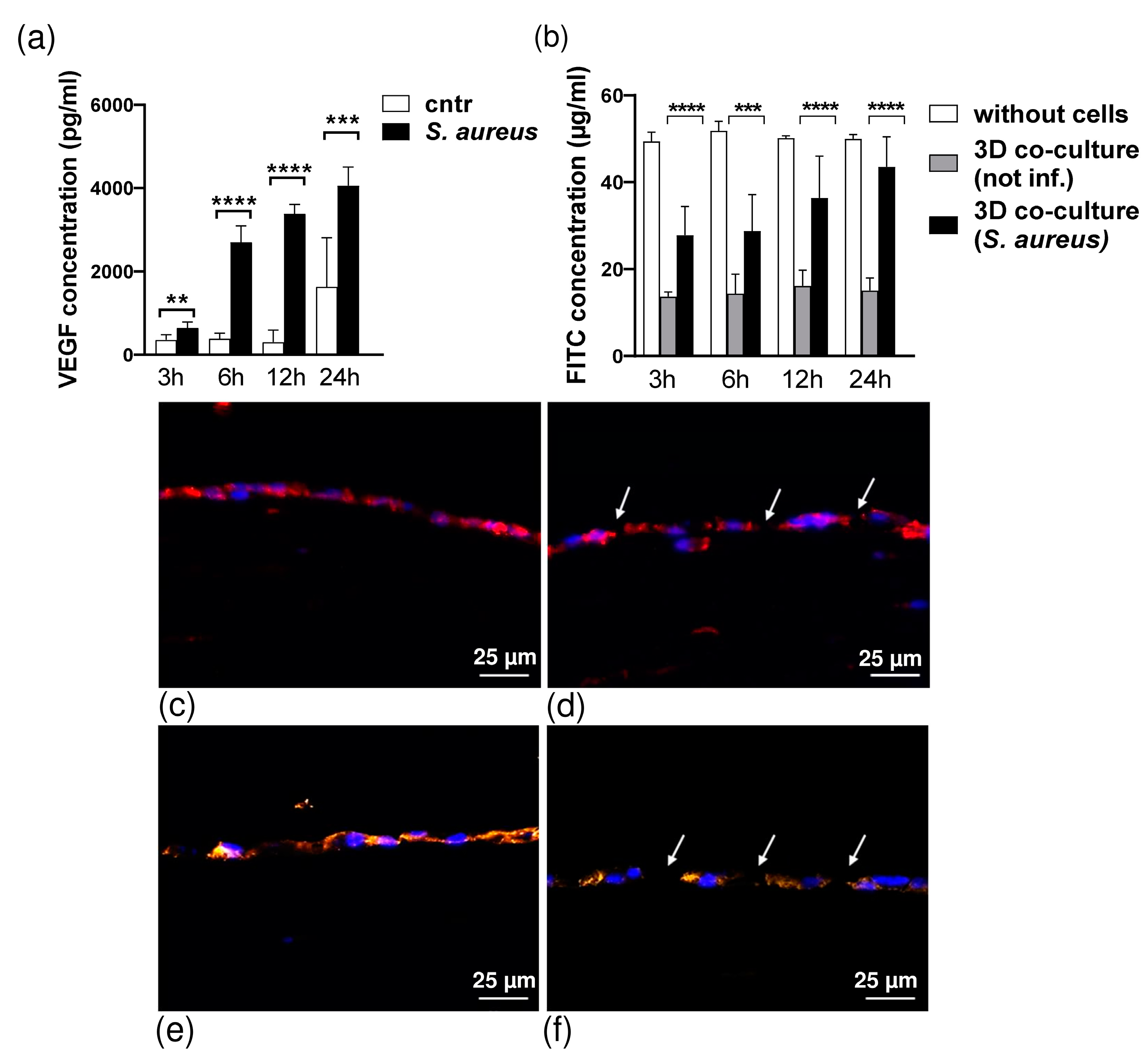
3.2. Cytokine Production in 3D Organotypic Co-Culture Model of Pleura after Infection
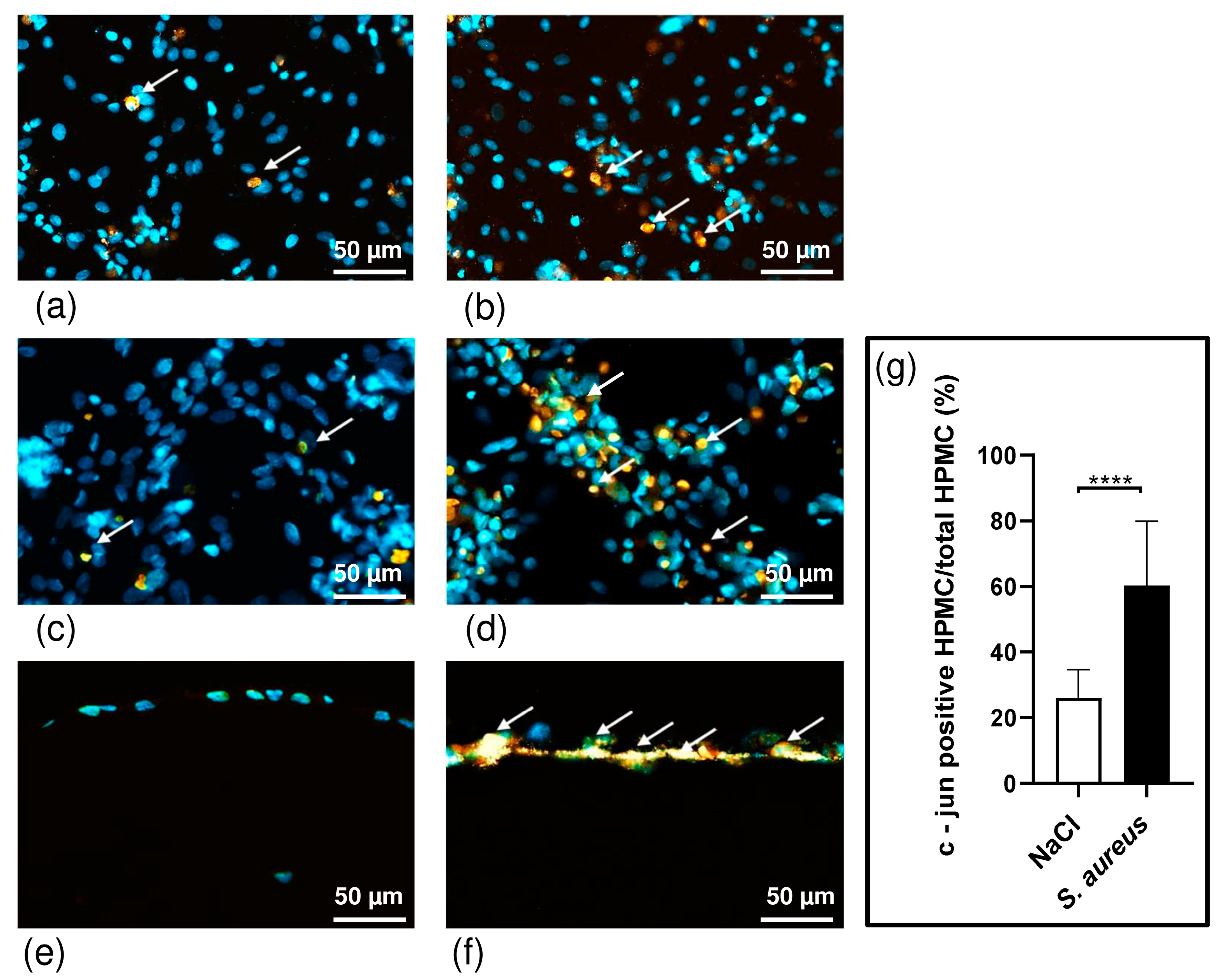
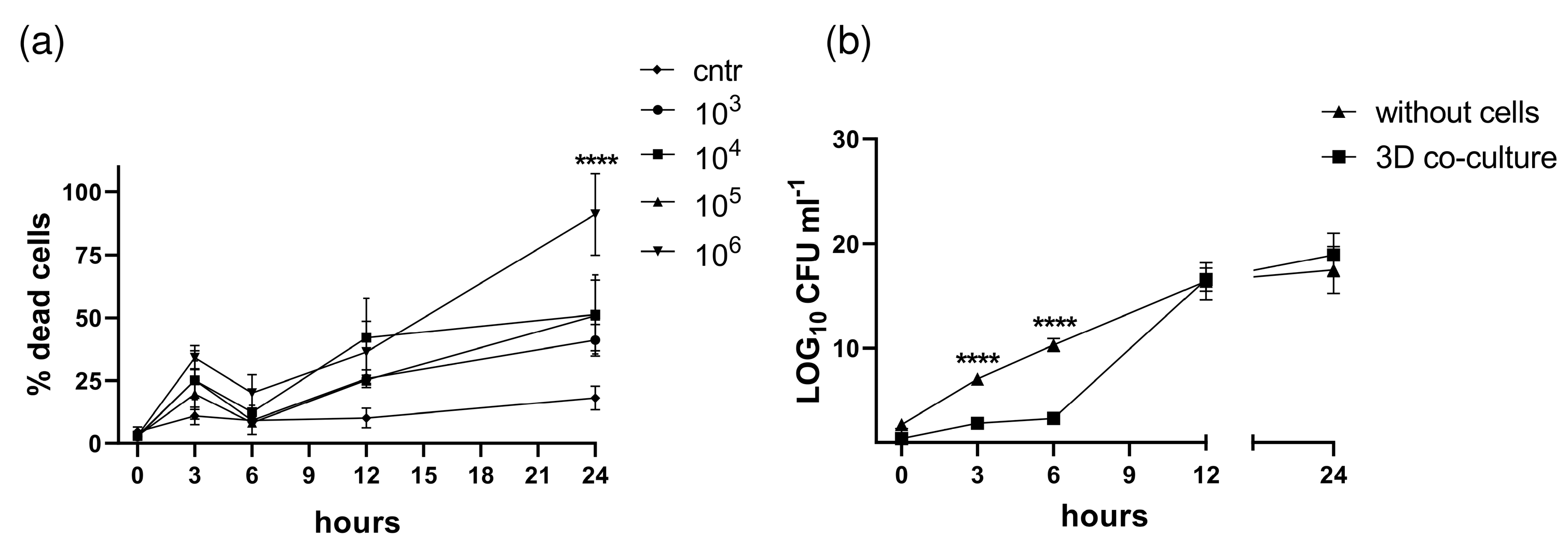
3.3. S. aureus Potently Induces Death of Pleural Fibroblasts and Mesothelial Cells Followed by Destruction of 3D Co-Culture Model of Pleura
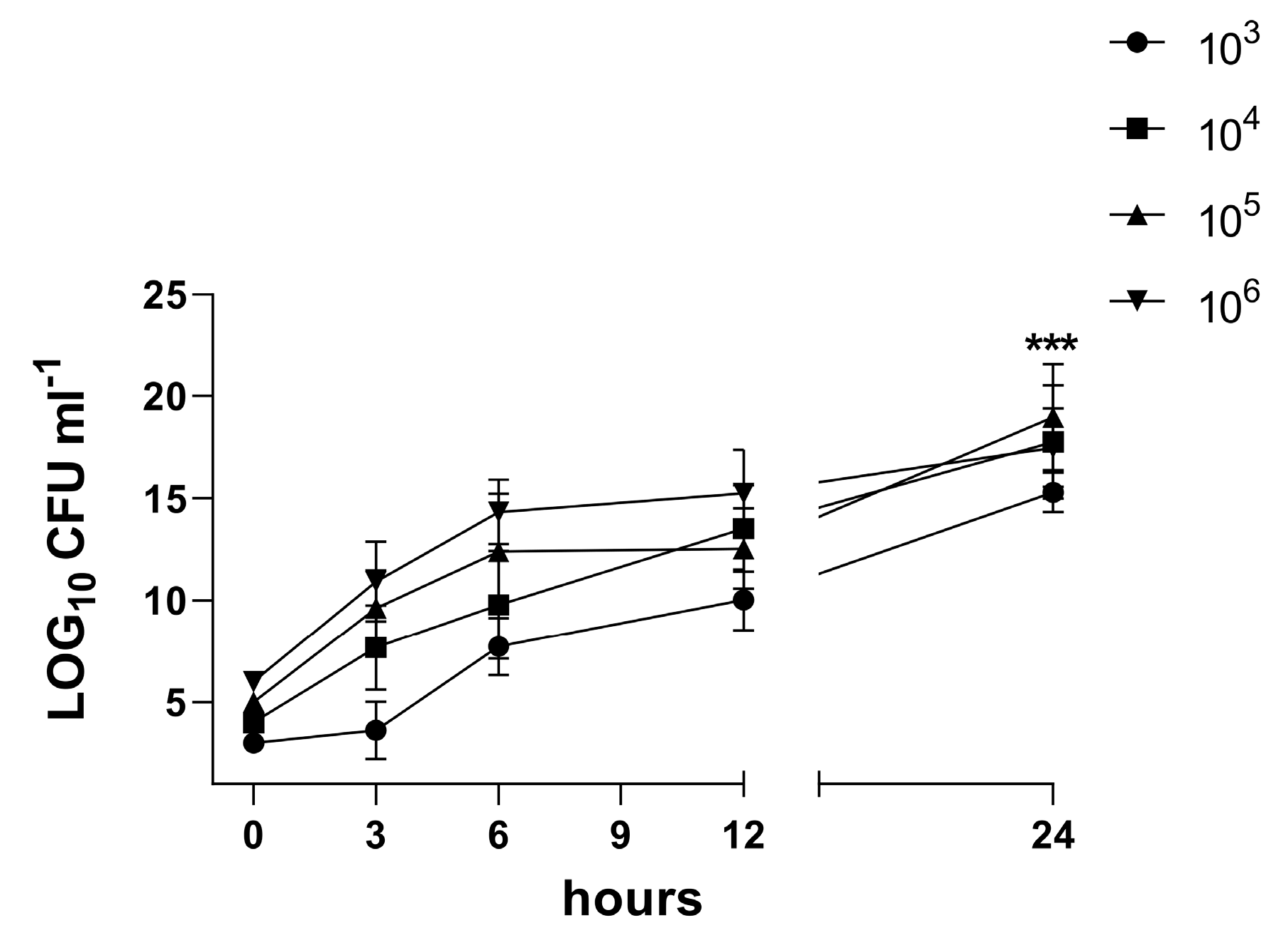
3.4. Characterization of the Biofilm in the 3D Co-Culture Model of Pleura
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassan, M.; Cargill, T.; Harriss, E.; Asciak, R.; Mercer, R.M.; Bedawi, E.O.; McCracken, D.J.; Psallidas, I.; Corcoran, J.P.; Rahman, N.M. The microbiology of pleural infection in adults: A systematic review. Eur. Respir. J. 2019, 54, 1900542. [Google Scholar] [CrossRef] [PubMed]
- Cargill, T.N.; Hassan, M.; Corcoran, J.P.; Harriss, E.; Asciak, R.; Mercer, R.M.; McCracken, D.J.; Bedawi, E.O.; Rahman, N.M. A systematic review of comorbidities and outcomes of adult patients with pleural infection. Eur. Respir. J. 2019, 54, 1900541. [Google Scholar] [CrossRef] [PubMed]
- Bedawi, E.O.; Hassan, M.; McCracken, D.; Rahman, N.M. Pleural infection: A closer look at the etiopathogenesis, microbiology and role of antibiotics. Expert Rev. Respir. Med. 2019, 13, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Schaudinn, C.; Kusanovic, J.P.; Gorur, A.; Gotsch, F.; Webster, P.; Nhan-Chang, C.L.; Erez, O.; Kim, C.J.; Espinoza, J.; et al. Detection of a microbial biofilm in intraamniotic infection. Am. J. Obstet. Gynecol. 2008, 198, 135.e1–135.e5. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Anwar, H.; Strap, J.L.; Costerton, J.W. Kinetic interaction of biofilm cells of Staphylococcus aureus with cephalexin and tobramycin in a chemostat system. Antimicrob. Agents Chemother. 1992, 36, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Daum, R.S.; Spellberg, B. Progress toward a Staphylococcus aureus vaccine. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2012, 54, 560–567. [Google Scholar] [CrossRef]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef]
- DuMont, A.L.; Torres, V.J. Cell targeting by the Staphylococcus aureus pore-forming toxins: It’s not just about lipids. Trends Microbiol. 2014, 22, 21–27. [Google Scholar] [CrossRef]
- Spaan, A.N.; Vrieling, M.; Wallet, P.; Badiou, C.; Reyes-Robles, T.; Ohneck, E.A.; Benito, Y.; De Haas, C.J.; Day, C.J.; Jennings, M.P.; et al. The staphylococcal toxins γ-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat. Commun. 2014, 5, 5438. [Google Scholar] [CrossRef]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Gendre, D.A.; Ameti, E.; Karenovics, W.; Perriraz-Mayer, N.; Triponez, F.; Serre-Beinier, V. Optimization of tumor spheroid model in mesothelioma and lung cancers and anti-cancer drug testing in H2052/484 spheroids. Oncotarget 2021, 12, 2375–2387. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model. Mech. 2019, 12, dmm039347. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Charles, C.A.; Ricotti, C.A.; Davis, S.C.; Mertz, P.M.; Kirsner, R.S. Use of tissue-engineered skin to study in vitro biofilm development. Dermatol. Surg. 2009, 35, 1334–1341. [Google Scholar] [CrossRef]
- Nichols, J.E.; Niles, J.A.; Vega, S.P.; Argueta, L.B.; Eastaway, A.; Cortiella, J. Modeling the lung: Design and development of tissue engineered macro- and micro-physiologic lung models for research use. Exp. Biol. Med. 2014, 239, 1135–1169. [Google Scholar] [CrossRef]
- Chang, R.; Emami, K.; Wu, H.; Sun, W. Biofabrication of a three-dimensional liver micro-organ as an in vitro drug metabolism model. Biofabrication 2010, 2, 45004. [Google Scholar] [CrossRef]
- Grapin-Botton, A. Three-dimensional pancreas organogenesis models. Diabetes Obes. Metab. 2016, 18 (Suppl. S1), 33–40. [Google Scholar] [CrossRef]
- Kenny, H.A.; Dogan, S.; Zillhardt, M.; KMitra, A.; Yamada, S.D.; Krausz, T.; Lengyel, E. Organotypic models of metastasis: A three-dimensional culture mimicking the human peritoneum and omentum for the study of the early steps of ovarian cancer metastasis. Cancer Treat. Res. 2009, 149, 335–351. [Google Scholar] [PubMed]
- Shacham-Silverberg, V.; Wells, J.M. Generation of esophageal organoids and organotypic raft cultures from human pluripotent stem cells. Methods Cell Biol. 2020, 159, 1–22. [Google Scholar] [PubMed]
- Lewis-Israeli, Y.R.; Volmert, B.D.; Gabalski, M.A.; Huang, A.R.; Aguirre, A. Generating Self-Assembling Human Heart Organoids Derived from Pluripotent Stem Cells. J. Vis. Exp. 2021, 175, e63097. [Google Scholar]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Metelmann, I.B.; Kraemer, S.; Steinert, M.; Langer, S.; Stock, P.; Kurow, O. Novel 3D organotypic co-culture model of pleura. PLoS ONE 2022, 17, e0276978. [Google Scholar] [CrossRef]
- Schreiter, J.S.; Beescho, C.; Kang, J.; Kursawe, L.; Moter, A.; Kikhney, J.; Langer, S.; Osla, F.; Wellner, E.; Kurow, O. New model in diabetic mice to evaluate the effects of insulin therapy on biofilm development in wounds. GMS Interdiscip. Plast. Reconstr. Surg. DGPW 2020, 9, Doc06. [Google Scholar]
- Langer, S.; Beescho, C.; Ring, A.; Dorfmann, O.; Steinau, H.U.; Spindler, N. A new in vivo model using a dorsal skinfold chamber to investigate microcirculation and angiogenesis in diabetic wounds. GMS Interdiscip. Plast. Reconstr. Surg. DGPW 2016, 5, Doc09. [Google Scholar]
- Janssen, Y.M.; Matalon, S.; Mossman, B.T. Differential induction of c-fos, c-jun, and apoptosis in lung epithelial cells exposed to ROS or RNS. Am. J. Physiol. 1997, 273, L789–L796. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Bakiri, L.; Yaniv, M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997, 16, 1695–1709. [Google Scholar] [CrossRef]
- Nasreen, N.; Mohammed, K.A.; Sanders, K.L.; Hardwick, J.; Van Horn, R.D.; Sharma, R.K.; Kilani, M.; Antony, V.B. Pleural mesothelial cells modulate polymorphonuclear leukocyte apoptosis in empyema. J. Clin. Immunol. 2003, 23, 1–10. [Google Scholar] [CrossRef]
- Mohammed, K.A.; Nasreen, N.; Hardwick, J.; Logie, C.S.; Patterson, C.E.; Antony, V.B. Bacterial induction of pleural mesothelial monolayer barrier dysfunction. J. Physiol.—Lung Cell Mol. Physiol. 2001, 281, L119–L125. [Google Scholar] [CrossRef] [PubMed]
- Grijalva, C.G.; Zhu, Y.; Nuorti, J.P.; Griffin, M.R. Emergence of parapneumonic empyema in the USA. Thorax 2011, 66, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Finley, C.; Clifton, J.; Fitzgerald, J.M.; Yee, J. Empyema: An increasing concern in Canada. Can. Respir. J. 2008, 15, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, C.J.; Paull, D.E.; Moezzi, J.E.; Scott, R.P.; Anstadt, M.P.; York, V.V.; Little, A.G. Choice of first intervention is related to outcomes in the management of empyema. Ann. Thorac. Surg. 2009, 87, 1525–1531. [Google Scholar] [CrossRef]
- Lehtomäki, A.; Nevalainen, R.; Ukkonen, M.; Nieminen, J.; Laurikka, J.; Khan, J. Trends in the Incidence, Etiology, Treatment, and Outcomes of Pleural Infections in Adults Over a Decade in a Finnish University Hospital. Scand. J. Surg. 2020, 109, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Hage, C.A.; abdul-Mohammed, K.; Antony, V.B. Pathogenesis of pleural infection. Respirology 2004, 9, 12–15. [Google Scholar] [CrossRef]
- Haagen, I.A.; Heezius, H.C.; Verkooyen, R.P.; Verhoef, J.; Verbrugh, H.A. Adherence of peritonitis-causing staphylococci to human peritoneal mesothelial cell monolayers. J. Infect. Dis. 1990, 161, 266–273. [Google Scholar] [CrossRef]
- Nasreen, N.; Mohammed, K.A.; Dowling, P.A.; Ward, M.J.; Galffy, G.; Antony, V.B. Talc induces apoptosis in human malignant mesothelioma cells in vitro. Am. J. Respir. Crit. Care Med. 2000, 161 Pt 1, 595–600. [Google Scholar] [CrossRef]
- Mohammed, K.A.; Nasreen, N.; Ward, M.J.; Antony, V.B. Induction of acute pleural inflammation by Staphylococcus aureus. I. CD4+ T cells play a critical role in experimental empyema. J. Infect. Dis. 2000, 181, 1693–1699. [Google Scholar] [CrossRef]
- Murphy, J.; Ramezanpour, M.; Roscioli, E.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Mucosal zinc deficiency in chronic rhinosinusitis with nasal polyposis contributes to barrier disruption and decreases ZO-1. Allergy 2018, 73, 2095–2097. [Google Scholar] [CrossRef]
- Malik, Z.; Roscioli, E.; Murphy, J.; Ou, J.; Bassiouni, A.; Wormald, P.J.; Vreugde, S. Staphylococcus aureus impairs the airway epithelial barrier in vitro. Int. Forum Allergy Rhinol. 2015, 5, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Hon, K.; Bennett, C.; Hu, H.; Menberu, M.; Wormald, P.J.; Zhao, Y.; Vreugde, S.; Liu, S. Corynebacterium accolens inhibits Staphylococcus aureus induced mucosal barrier disruption. Front. Microbiol. 2022, 13, 984741. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, K.A.; Nasreen, N.; Antony, V.B. Bacterial induction of early response genes and activation of proapoptotic factors in pleural mesothelial cells. Lung 2007, 185, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.-T.; Peisl, L.; Barletta, F.; Luqman, A.; Götz, F. Toll-Like Receptor 2 and Lipoprotein-Like Lipoproteins Enhance Staphylococcus aureus Invasion in Epithelial Cells. Infect. Immun. 2018, 86, e00343-e18. [Google Scholar] [CrossRef] [PubMed]
- Eidels, L.; Proia, R.L.; Hart, D.A. Membrane receptors for bacterial toxins. Microbiol. Rev. 1983, 47, 596–620. [Google Scholar] [CrossRef]
- Wilkosz, S.; Edwards, L.A.; Bielsa, S.; Hyams, C.; Taylor, A.; Davies, R.J.; Laurent, G.J.; Chambers, R.C.; Brown, J.S. Characterization of a new mouse model of empyema and the mechanisms of pleural invasion by Streptococcus pneumoniae. Am. J. Respir. Cell Mol. Biol. 2012, 46, 180–187. [Google Scholar] [CrossRef]
- Glading, A.; Bodnar, R.J.; Reynolds, I.J.; Shiraha, H.; Satish, L.; Potter, D.A.; Blair, H.C.; Wells, A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol. Cell. Biol. 2004, 24, 2499–2512. [Google Scholar] [CrossRef]
- Roberts, W.G.; Palade, G.E. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J. Cell Sci. 1995, 108 Pt 6, 2369–2379. [Google Scholar] [CrossRef]
- Thickett, D.R.; Armstrong, L.; Millar, A.B. Vascular endothelial growth factor (VEGF) in inflammatory and malignant pleural effusions. Thorax 1999, 54, 707–710. [Google Scholar] [CrossRef]
- Becker, P.M.; Alcasabas, A.; Yu, A.Y.; Semenza, G.L.; Bunton, T.E. Oxygen-independent upregulation of vascular endothelial growth factor and vascular barrier dysfunction during ventilated pulmonary ischemia in isolated ferret lungs. Am. J. Respir. Cell Mol. Biol. 2000, 22, 272–279. [Google Scholar] [CrossRef]
- Ishimoto, O.; Saijo, Y.; Narumi, K.; Kimura, Y.; Ebina, M.; Matsubara, N.; Asou, N.; Nakai, Y.; Nukiwa, T. High level of vascular endothelial growth factor in hemorrhagic pleural effusion of cancer. Oncology 2002, 63, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.G.; Melkerneker, D.; Thompson, P.J.; Light, R.W.; Lane, K.B. Transforming growth factor beta induces vascular endothelial growth factor elaboration from pleural mesothelial cells in vivo and in vitro. Am. J. Respir. Crit. Care Med. 2002, 165, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Gallagher, E. From JNK to pay dirt: Jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 2005, 57, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, pages E131–E136. [Google Scholar] [CrossRef]
- Dong, Z.; Crawford, H.C.; Lavrovsky, V.; Taub, D.; Watts, R.; Matrisian, L.M.; Colburn, N.H. A dominant negative mutant of jun blocking 12-O-tetradecanoylphorbol-13-acetate-induced invasion in mouse keratinocytes. Mol. Carcinog. 1997, 19, 204–212. [Google Scholar] [CrossRef]
- Lembke, C.; Podbielski, A.; Hidalgo-Grass, C.; Jonas, L.; Hanski, E.; Kreikemeyer, B. Characterization of biofilm formation by clinically relevant serotypes of group A streptococci. Appl. Environ. Microbiol. 2006, 72, 2864–2875. [Google Scholar] [CrossRef]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]
- Thacker, V.V.; Dhar, N.; Sharma, K.; Barrile, R.; Karalis, K.; McKinney, J.D. A lung-on-chip model of early Mycobacterium tuberculosis infection reveals an essential role for alveolar epithelial cells in controlling bacterial growth. eLife 2020, 9, e59961. [Google Scholar] [CrossRef]
- Ozdogan, C.Y.; Kenar, H.; Davun, K.E.; Yucel, D.; Doger, E.; Alagoz, S. An in vitro 3D diabetic human skin model from diabetic primary cells. Biomed. Mater. 2020, 16, 15027. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Incubation Period (Hours) | Group B: S. aureus (1 × 103 CFU/mL) | Group C: S. aureus (1 × 106 CFU/mL) |
|---|---|---|
| 0 | no bacteria | no bacteria |
| 3 | single bacterial cells | bacterial micro-colonies |
| 6 | bacterial micro-colonies | bacterial aggregates |
| 12 | bacterial aggregates in tissue, biofilm | mature biofilm |
| 24 | mature biofilm | mature biofilm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurow, O.; Nuwayhid, R.; Stock, P.; Steinert, M.; Langer, S.; Krämer, S.; Metelmann, I.B. Organotypic 3D Co-Culture of Human Pleura as a Novel In Vitro Model of Staphylococcus aureus Infection and Biofilm Development. Bioengineering 2023, 10, 537. https://doi.org/10.3390/bioengineering10050537
Kurow O, Nuwayhid R, Stock P, Steinert M, Langer S, Krämer S, Metelmann IB. Organotypic 3D Co-Culture of Human Pleura as a Novel In Vitro Model of Staphylococcus aureus Infection and Biofilm Development. Bioengineering. 2023; 10(5):537. https://doi.org/10.3390/bioengineering10050537
Chicago/Turabian StyleKurow, Olga, Rima Nuwayhid, Peggy Stock, Matthias Steinert, Stefan Langer, Sebastian Krämer, and Isabella B. Metelmann. 2023. "Organotypic 3D Co-Culture of Human Pleura as a Novel In Vitro Model of Staphylococcus aureus Infection and Biofilm Development" Bioengineering 10, no. 5: 537. https://doi.org/10.3390/bioengineering10050537