
Assessment of the Performances of the Protein Modeling Techniques Participating in CASP15 Using a Structure-Based Functional Site Prediction Approach: ResiRole

Abstract
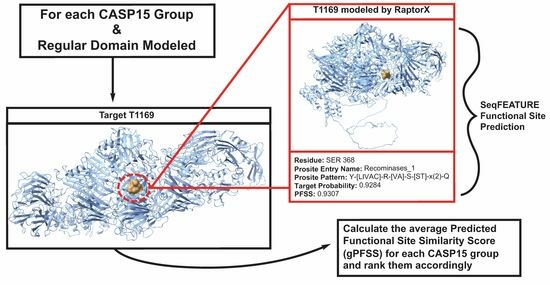
:
1. Introduction
2. Materials and Methods
2.1. Calculation of Quality Metrics: The Average and Cumulative PFSSs
2.2. Description of the CASP15 Dataset Used in the Study
3. Results
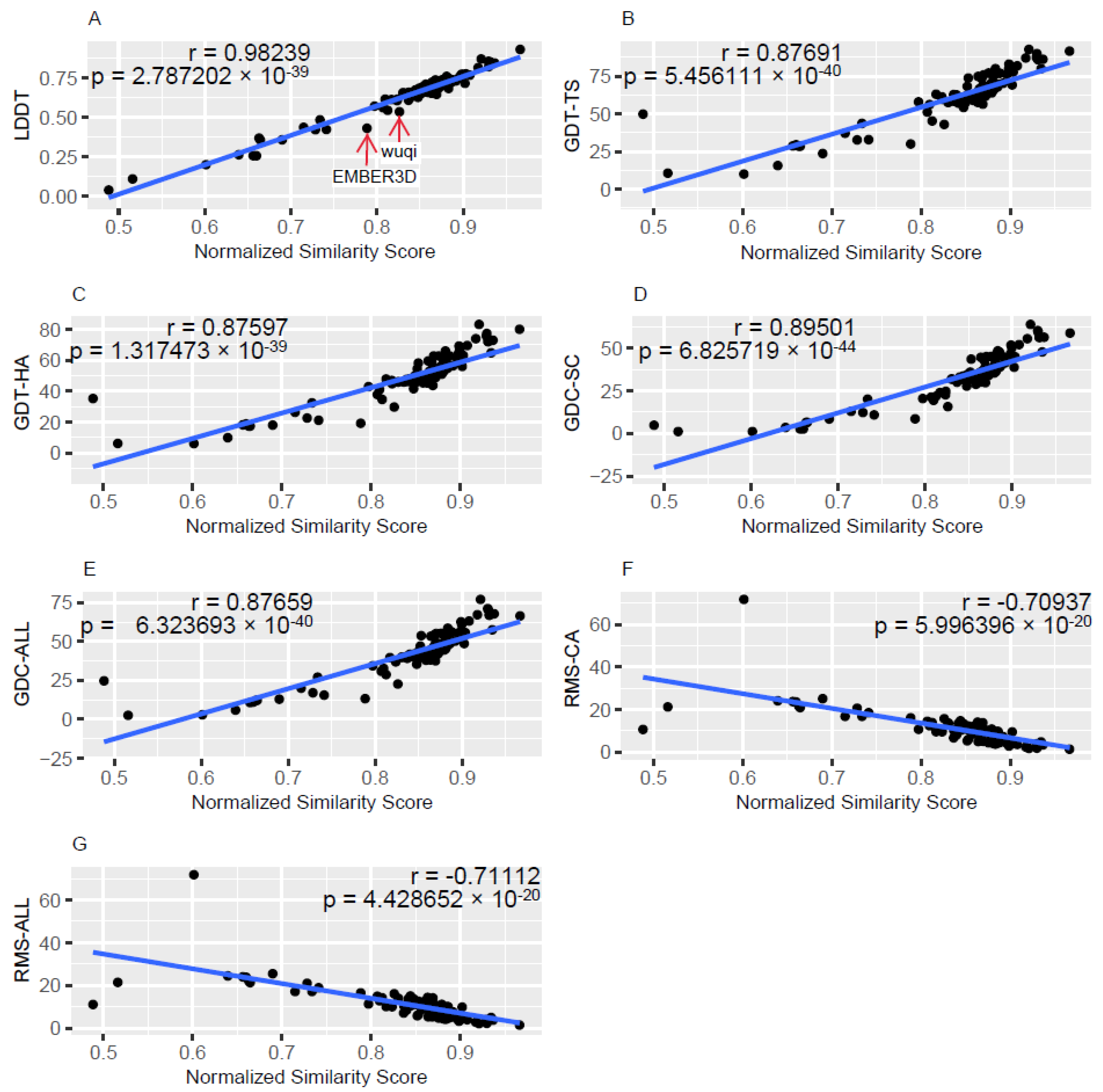
3.1. Description of the Relationships between gPFSS and Standard Quality Metrics
3.2. Ranking Modeling Groups Using the gPFSS and cumPFSS Performance Metrics
3.3. Statistical Comparisons of the Modeling Group Performances
3.4. Illustrative Examples of the Utility of the PFSS Metric
4. Discussion
4.1. Comparison of gPFSS to Standard Metrics
4.2. Further Descriptions of Individual Group Performances
4.3. Applications for Guiding Functional Characterizations
4.4. Limitations of the Described Methodology
4.5. Possible Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carugo, O.; Pongor, S. A normalized root-mean-spuare distance for comparing protein three-dimensional structures. Protein Sci. 2001, 10, 1470–1473. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003, 31, 3370–3374. [Google Scholar] [CrossRef] [PubMed]
- Olechnovič, K.; Kulberkytė, E.; Venclovas, Č. CAD-score: A new contact area difference-based function for evaluation of protein structural models. Proteins Struct. Funct. Bioinform. 2013, 81, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Antczak, P.L.M.; Ratajczak, T.; Lukasiak, P.; Blazewicz, J. SphereGrinder-reference structure-based tool for quality assessment of protein structural models. In Proceedings of the 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Washington, DC, USA, 9–12 November 2015; pp. 665–668. [Google Scholar]
- Olechnovič, K.; Monastyrskyy, B.; Kryshtafovych, A.; Venclovas, Č. Comparative analysis of methods for evaluation of protein models against native structures. Bioinformatics 2019, 35, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Croll, T.I.; Sammito, M.D.; Kryshtafovych, A.; Read, R.J. Evaluation of template-based modeling in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1113–1127. [Google Scholar] [CrossRef]
- Kwon, S.; Won, J.; Kryshtafovych, A.; Seok, C. Assessment of protein model structure accuracy estimation in CASP14: Old and new challenges. Proteins 2021, 89, 1940–1948. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Guo, Z.; Wu, T.; Hou, J.; Cheng, J. Protein model accuracy estimation empowered by deep learning and inter-residue distance prediction in CASP14. Sci. Rep. 2021, 11, 10943. [Google Scholar] [CrossRef]
- Haas, J.; Gumienny, R.; Barbato, A.; Ackermann, F.; Tauriello, G.; Bertoni, M.; Studer, G.; Smolinski, A.; Schwede, T. Introducing “best single template” models as reference baseline for the Continuous Automated Model Evaluation (CAMEO). Proteins Struct. Funct. Bioinform. 2019, 87, 1378–1387. [Google Scholar] [CrossRef]
- Toth, J.M.; DePietro, P.J.; Haas, J.; McLaughlin, W.A. ResiRole: Residue-level functional site predictions to gauge the accuracies of protein structure prediction techniques. Bioinformatics 2021, 37, 351–359. [Google Scholar] [CrossRef]
- Wu, S.; Liang, M.P.; Altman, R.B. The SeqFEATURE library of 3D functional site models: Comparison to existing methods and applications to protein function annotation. Genome Biol. 2008, 9, 1. [Google Scholar] [CrossRef]
- Kinch, L.N.; Schaeffer, R.D.; Kryshtafovych, A.; Grishin, N.V. Target classification in the 14th round of the critical assessment of protein structure prediction (CASP14). Proteins Struct. Funct. Bioinform. 2021, 89, 1618–1632. [Google Scholar] [CrossRef]
- Elofsson, A. Progress at protein structure prediction, as seen in CASP15. Curr. Opin. Struct. Biol. 2023, 80, 102594. [Google Scholar] [CrossRef]
- Van Rossum, G. Python Programming Language. In Proceedings of the USENIX Annual Technical Conference, Santa Clara, CA, USA, 17–22 June 2007; Volume 41, pp. 1–36. [Google Scholar]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K. CASP prediction center infrastructure and evaluation measures in CASP10 and CASP ROLL. Proteins Struct. Funct. Bioinform. 2014, 82, 7–13. [Google Scholar] [CrossRef]
- Hulo, N.; Bairoch, A.; Bulliard, V.; Cerutti, L.; De Castro, E.; Langendijk-Genevaux, P.S.; Pagni, M.; Sigrist, C.J. The PROSITE database. Nucleic Acids Res. 2006, 34 (Suppl. 1), D227–D230. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2017. Available online: https://www.R-project.org (accessed on 16 October 2023).
- Huber, F. A Logical Introduction to Probability and Induction; Oxford University Press: New York, NY, USA, 2018. [Google Scholar]
- Weissenow, K.; Heinzinger, M.; Steinegger, M.; Rost, B. Ultra-fast protein structure prediction to capture effects of sequence variation in mutation movies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K. CASP 11 statistics and the prediction center evaluation system. Proteins Struct. Funct. Bioinform. 2016, 84, 15–19. [Google Scholar] [CrossRef]
- Simpkin, A.J.; Mesdaghi, S.; Sánchez Rodríguez, F.; Elliott, L.; Murphy, D.L.; Kryshtafovych, A.; Keegan, R.M.; Rigden, D.J. Tertiary structure assessment at CASP15. Proteins Struct. Funct. Bioinform. 2023, 91, 1616–1635. [Google Scholar] [CrossRef]
- Oda, T. Improving Protein Structure Prediction with Extended Sequence Similarity Searches and Deep-Learning-Based Refinement in CASP15. Proteins 2023, 91, 1712–1723. [Google Scholar] [CrossRef]
- Joosten, R.P.; Te Beek, T.A.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.; Schneider, R.; Sander, C.; Vriend, G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2010, 39 (Suppl. 1), D411–D419. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Liu, S.; Xia, X.; Calvo, E.; Zhou, Z.H. Native structure of mosquito salivary protein uncovers domains relevant to pathogen transmission. Nat. Commun. 2023, 14, 899. [Google Scholar] [CrossRef]
- Biasini, M.; Schmidt, T.; Bienert, S.; Mariani, V.; Studer, G.; Haas, J.; Johner, N.; Schenk, A.D.; Philippsen, A.; Schwede, T. OpenStructure: An integrated software framework for computational structural biology. Acta Crystallogr. D Biol. Crystallogr. 2013, 69 Pt 5, 701–709. [Google Scholar] [CrossRef]
- Fowle, H.; Zhao, Z.; Grana, X. PP2A holoenzymes, substrate specificity driving cellular functions and deregulation in cancer. Adv. Cancer Res. 2019, 144, 55–93. [Google Scholar] [CrossRef]
- Pourmal, S.; Green, E.; Bajaj, R.; Chemmama, I.; Knudsen, G.; Gupta, M.; Sali, A.; Cheng, Y.; Craik, C.; Kroetz, D. Structural basis of prostaglandin efflux by MRP4. bioRxiv 2022. [Google Scholar] [CrossRef]
- Jing, X.; Xu, J. Fast and effective protein model refinement using deep graph neural networks. Nat. Comput. Sci. 2021, 1, 462–469. [Google Scholar] [CrossRef]
- Dawson, N.L.; Lewis, T.E.; Das, S.; Lees, J.G.; Lee, D.; Ashford, P.; Orengo, C.A.; Sillitoe, I. CATH: An expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017, 45, D289–D295. [Google Scholar] [CrossRef]
- Ahdritz, G.; Bouatta, N.; Kadyan, S.; Xia, Q.; Gerecke, W.; O’Donnell, T.J.; Berenberg, D.; Fisk, I.; Zanichelli, N.; Zhang, B. OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. bioRxiv 2022. [Google Scholar] [CrossRef]
- Sillitoe, I.; Bordin, N.; Dawson, N.; Waman, V.P.; Ashford, P.; Scholes, H.M.; Pang, C.S.; Woodridge, L.; Rauer, C.; Sen, N. CATH: Increased structural coverage of functional space. Nucleic Acids Res. 2021, 49, D266–D273. [Google Scholar] [CrossRef]
- Varadi, M.; Velankar, S. The impact of AlphaFold Protein Structure Database on the fields of life sciences. Proteomics 2023, 23, 2200128. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Group Name | cumPFSS | gPFSS | Average GDT-TS | Domains |
|---|---|---|---|---|
| UM-TBM | 89.1575 | 0.9098 ± 0.069 | 83.8797 ± 16.125 | 98 |
| Yang_Server | 89.0518 | 0.9087 ± 0.069 | 84.2358 ± 15.183 | 98 |
| Yang | 88.842 | 0.9066 ± 0.068 | 83.3322 ± 17.207 | 98 |
| MULTICOM_refine | 88.114 | 0.8991 ± 0.086 | 79.012 ± 21.53 | 98 |
| MULTICOM_qa | 88.0744 | 0.8987 ± 0.086 | 78.4293 ± 21.723 | 98 |
| Kiharalab | 88.0307 | 0.8983 ± 0.086 | 79.5234 ± 19.03 | 98 |
| MULTICOM_deep | 87.9948 | 0.8979 ± 0.087 | 78.6633 ± 21.771 | 98 |
| MULTICOM | 87.9889 | 0.8978 ± 0.082 | 80.1332 ± 20.214 | 98 |
| RaptorX | 87.9075 | 0.897 ± 0.08 | 77.5924 ± 21.971 | 98 |
| MULTICOM_egnn | 87.871 | 0.8966 ± 0.087 | 78.6379 ± 21.508 | 98 |
| server_126 | 87.851 | 0.8964 ± 0.08 | 76.6975 ± 22.13 | 98 |
| BAKER | 87.8253 | 0.8962 ± 0.072 | 78.9276 ± 19.247 | 98 |
| GuijunLab_DeepDA | 87.5177 | 0.893 ± 0.09 | 76.0575 ± 22.965 | 98 |
| McGuffin | 87.5162 | 0.893 ± 0.089 | 79.6776 ± 21.1 | 98 |
| Bhattacharya | 87.4844 | 0.8927 ± 0.089 | 72.7336 ± 23.766 | 98 |
| GuijunLab_Threader | 87.4766 | 0.8926 ± 0.092 | 75.4201 ± 23.506 | 98 |
| MUFold | 87.4764 | 0.8926 ± 0.088 | 75.7452 ± 23.314 | 98 |
| server_124 | 87.4566 | 0.8924 ± 0.081 | 76.6425 ± 22.425 | 98 |
| Manifold | 87.4383 | 0.8922 ± 0.092 | 78.3286 ± 21.427 | 98 |
| Agemo_mix | 87.4285 | 0.8921 ± 0.092 | 75.4971 ± 24.464 | 98 |
| Manifold_E | 87.3873 | 0.8917 ± 0.092 | 78.4974 ± 21.096 | 98 |
| DFolding | 87.2864 | 0.8907 ± 0.088 | 79.921 ± 21.185 | 98 |
| DFolding_server | 87.2661 | 0.8905 ± 0.089 | 77.5 ± 23.235 | 98 |
| AP_1 | 87.2457 | 0.8903 ± 0.078 | 72.0505 ± 25.516 | 98 |
| bench | 87.2254 | 0.8992 ± 0.078 | 79.1023 ± 19.739 | 97 |
| Group Name | cumPFSS | gPFSS | Average GDT-TS | Domains |
|---|---|---|---|---|
| Yang | 34.9172 | 0.8953 ± 0.079 | 76.1839 ± 21.087 | 39 |
| UM-TBM | 34.885 | 0.8945 ± 0.078 | 77.771 ± 19.278 | 39 |
| bench | 34.3215 | 0.88 ± 0.094 | 70.311 ± 24.287 | 39 |
| Kiharalab | 34.2139 | 0.8773 ± 0.106 | 70.85 ± 23.167 | 39 |
| MULTICOM | 34.0653 | 0.8735 ± 0.102 | 69.8126 ± 24.293 | 39 |
| MULTICOM_refine | 34.0649 | 0.8735 ± 0.107 | 67.9574 ± 26.441 | 39 |
| BAKER | 34.0433 | 0.8729 ± 0.092 | 69.1556 ± 21.431 | 39 |
| Yang_Server | 33.9965 | 0.8946 ± 0.083 | 79.6832 ± 17.871 | 38 |
| MULTICOM_qa | 33.9227 | 0.8698 ± 0.107 | 67.4618 ± 26.177 | 39 |
| server_126 | 33.9147 | 0.8696 ± 0.097 | 63.9297 ± 26.457 | 39 |
| MULTICOM_egnn | 33.9086 | 0.8695 ± 0.109 | 68.0495 ± 26.275 | 39 |
| Shennong | 33.8923 | 0.869 ± 0.104 | 65.7933 ± 25.358 | 39 |
| Wallner | 33.8653 | 0.8683 ± 0.106 | 66.4941 ± 28.466 | 39 |
| McGuffin | 33.7385 | 0.8651 ± 0.103 | 69.0397 ± 23.96 | 39 |
| DFolding | 33.7349 | 0.865 ± 0.109 | 70.3521 ± 24.097 | 39 |
| RaptorX | 33.7334 | 0.865 ± 0.098 | 65.0223 ± 26.525 | 39 |
| server_124 | 33.6896 | 0.8638 ± 0.103 | 63.47 ± 27.09 | 39 |
| MUFold_H | 33.6521 | 0.8629 ± 0.103 | 63.9087 ± 27.151 | 39 |
| Bhattacharya | 33.621 | 0.8621 ± 0.108 | 59.8569 ± 27.209 | 39 |
| AP_1 | 33.5958 | 0.8614 ± 0.091 | 56.4226 ± 28.276 | 39 |
| Asclepius | 33.5758 | 0.8609 ± 0.11 | 63.1954 ± 28.892 | 39 |
| Manifold | 33.5282 | 0.8597 ± 0.117 | 66.6654 ± 26.378 | 39 |
| DFolding_server | 33.5281 | 0.8597 ± 0.108 | 65.7005 ± 28.139 | 39 |
| PEZYFoldings | 33.5064 | 0.8817 ± 0.095 | 74.9637 ± 22.453 | 38 |
| hFold_human | 33.4941 | 0.8588 ± 0.112 | 64.0833 ± 27.371 | 39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, G.J.; Parry, T.K.; McLaughlin, W.A. Assessment of the Performances of the Protein Modeling Techniques Participating in CASP15 Using a Structure-Based Functional Site Prediction Approach: ResiRole. Bioengineering 2023, 10, 1377. https://doi.org/10.3390/bioengineering10121377
Huang GJ, Parry TK, McLaughlin WA. Assessment of the Performances of the Protein Modeling Techniques Participating in CASP15 Using a Structure-Based Functional Site Prediction Approach: ResiRole. Bioengineering. 2023; 10(12):1377. https://doi.org/10.3390/bioengineering10121377
Chicago/Turabian StyleHuang, Geoffrey J., Thomas K. Parry, and William A. McLaughlin. 2023. "Assessment of the Performances of the Protein Modeling Techniques Participating in CASP15 Using a Structure-Based Functional Site Prediction Approach: ResiRole" Bioengineering 10, no. 12: 1377. https://doi.org/10.3390/bioengineering10121377